

H2-Generation from Alcohols by the MOF-Based Noble Metal-Free Photocatalyst Ni/CdS/TiO2@MIL-101*
Dominic Tilgner A , Mara Klarner A , Sebastian Hammon B , Martin Friedrich A , Andreas Verch C D , Niels de Jonge C E , Stephan Kümmel B and Rhett Kempe A F
A F
A Inorganic Chemistry II, Catalyst Design, University of Bayreuth, Bayreuth 95440, Germany.
B Theoretical Physics IV, University of Bayreuth, Bayreuth 95440, Germany.
C INM – Leibniz Institute for New Materials, Saarbrücken 66123, Germany.
D Carl Zeiss AG, Oberkochen 73447, Germany.
E Department of Physics, Saarland University, Saarbrücken 66123, Germany.
F Corresponding author. Email: kempe@uni-bayreuth.de
Australian Journal of Chemistry 72(10) 842-847 https://doi.org/10.1071/CH19255
Submitted: 4 June 2019 Accepted: 22 July 2019 Published: 15 August 2019
Abstract
The synthesis of important classes of chemical compounds from alcohols helps to conserve Earth’s fossil carbon resources, since alcohols can be obtained from indigestible and abundantly available biomass. The utilisation of visible light for the activation of alcohols permits alcohol-based C–N and C–C bond formation under mild conditions inaccessible with thermally operating hydrogen liberation catalysts. Herein, we report on a noble metal-free photocatalyst able to split alcohols into hydrogen and carbonyl compounds under inert gas atmosphere without the requirement of electron donors, additives, or aqueous reaction media. The reusable photocatalyst mediates C–N multiple bond formation using the oxidation of alcohols and subsequent coupling with amines. The photocatalyst consists of a CdS/TiO2 heterojunction decorated with co-catalytic Ni nanoparticles and is prepared on size-optimised colloidal metal–organic framework (MOF) crystallites.
The dehydrogenation of alcohols is a fundamental reaction in sustainable chemistry.[1] Alcohols can be obtained via pyrolysis and hydrogenation from lignocellulose,[2] an abundantly available, indigestible, and barely used biomass.[3] Thus, alcohols are a promising renewable carbon feedstock and the development of alcohol refunctionalisation reactions is the key to its use.[4] Alcohols are rather unreactive and dehydrogenation is an elegant way of activating them. The oxidised form, carbonyl compounds, can undergo condensation reactions permitting the formation of C–C or C–N multiple bonds. The synthesis of imines from alcohols and amines introduced by Milstein and co-workers is an example of broad interest for such a C–N bond formation reactions.[5] Our group has developed the synthesis of aromatic N-heterocyclic compounds from alcohols in which C–N and C–C multiple bond formation takes place concertedly.[4,6–9] We expected that a photocatalyst able to mediate C–N bond formation via dehydrogenative coupling under very mild reaction conditions is highly desirable to significantly extend the applicability of existing thermally operating noble metal-free catalysts for such reactions.[5b,6b,8b-c,10]
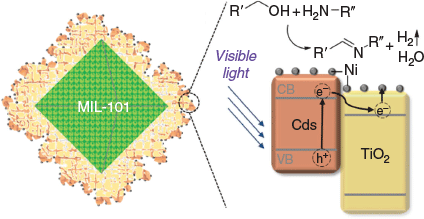
Here, we report on a photocatalyst that mediates visible light-driven imine synthesis from alcohols and amines. This C–N multiple bond formation takes place via dehydrogenation of alcohols under very mild conditions without sacrificial electron donors (alcohol splitting). Our noble metal-free photocatalyst consists of a CdS/TiO2 heterojunction prepared on a porous support that is decorated with co-catalytic Ni nanoparticles. The metal–organic framework (MOF) MIL-101 (Cr)[11] was used as the porous support material stabilising the photocatalytic system.[12] We demonstrate the acceptorless dehydrogenation of alcohols by splitting into hydrogen and carbonyl compounds at room temperature with a broad substrate scope to generate aldehydes and dialkyl, diaryl, and aryl-alkyl ketones. Visible light-driven alcohol dehydrogenation without electron donors is rarely found in the literature[13] and often requires aqueous media, including additives.[14] Aqueous media are disadvantageous regarding condensation steps, which are needed in many dehydrogenative coupling/condensation reactions.
We expected that the arrangement of CdS and TiO2 nanocrystals on a suitable MOF would be an elegant way to create heterojunction systems with efficient charge carrier separation and high photocatalytic activity. The pores of the MOF can be selectively loaded with precursor molecules to control the size and amount of the different nanocrystals without applying surface blocking ligands. MIL-101 (Cr) was chosen because of its stability during solvothermal modifications and its large pore volume. It was prepared according to a procedure described previously with an average crystallite size of 300 nm (Fig. S1, Supplementary Material).[15] This crystallite size is optimal regarding catalytic performance. The crystallites are large enough to ensure efficient separation for reusability and small enough to have a sufficiently large outer surface for modification with light harvesting materials. We recently reported a synthesis protocol of crystalline TiO2 (anatase) on the surface of MIL-101 crystallites leading to a core–shell morphology.[12] Titanium(iv) isopropoxide [Ti(OiPr)4)] was infiltrated into the pores of MIL-101 by gas-phase loading. The subsequent hydrolysis of the Ti precursor led to the formation of amorphous TiO2 inside the cavities of MIL-101. The crystallisation of TiO2 (anatase) on the surface of MOF crystallites was performed under hydrothermal conditions. The MIL-101 core is crucial since the material collapses by removing the support of the photocatalytic system.[12] In this work, the resulting TiO2@MIL-101 material was modified with CdS nanocrystals under solvothermal conditions using cadmium acetate dihydrate and dimethyl sulfoxide as precursors to yield CdS/TiO2@MIL-101.[16] MIL-101 serves as structure-directing centre to form a dense photoactive shell. Thereby, the MOF core offers a platform for the growth of CdS and TiO2 nanocrystallites covering the outer surface of single MIL-101 crystallites. Finally, we decorated the core–shell material with homogeneously distributed nickel nanoparticles using bis(cyclopentadienyl)nickel(ii) [Ni(Cp)2] as the volatile Ni precursor. The Ni/CdS/TiO2@MIL-101 photocatalyst and the process of charge carrier separation between the semiconducting materials is illustrated in Scheme 1. The TiO2 and CdS particles form a type II heterojunction at their interface where CdS serves as light absorbing component for the visible spectrum. The directed electron transfer from the conduction band of CdS into that of TiO2 is a well understood process.[17] We believe that the enhanced photocatalytic efficiency of a MOF-supported CdS/TiO2 heterojunction is due to a reduced charge recombination rate as described in the literature. Metallic Ni nanoparticles promote the separation of charges, since the electrical potential gradient at the Ni/semiconductor interface enhances the electron transfer.[18] Consequently, the charge carriers initiate spatially separated redox reactions, since they are distributed over three different components. The nanoparticles of the non-noble metal Ni act as electron reservoirs and, thus, as a co-catalyst for H2 liberation.
|
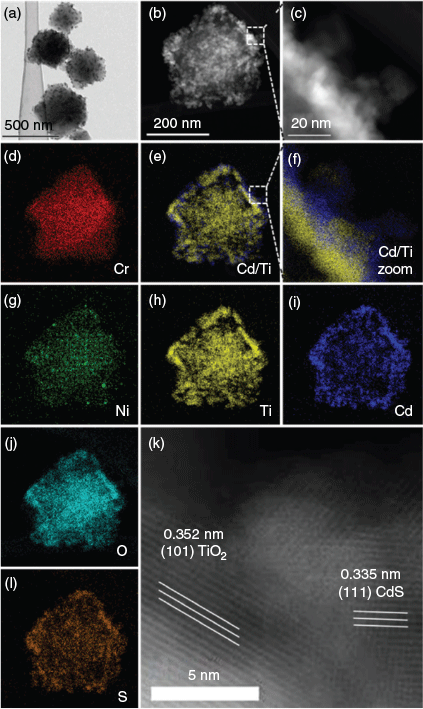
The initial specific surface area of MIL-101 decreased perceptibly during the generation of TiO2 and CdS, as investigated by nitrogen physisorption measurements (Fig. S2, Supplementary Material). Only a minor reduction of the specific surface area was observed after the modification with nickel. Crystalline TiO2 (anatase) and CdS (cubic) were identified by X-ray powder diffraction (XRD) analysis (Fig. S3, Supplementary Material). The characteristic reflexes between 2° and 20° (2θ) indicate the preserved structure of MIL-101. X-Ray photoelectron spectroscopy (XPS) analysis confirmed the presence of TiO2 and CdS. Signals of Ni0 and NiII were observed in the Ni 2p spectrum. The presence of NiII can be attributed to the formation of nickel(ii) hydroxide when exposed to air during sample handling (Fig. S4, Supplementary Material). We determined the elemental composition of the materials by inductively coupled plasma–optical emission spectrometry (ICP-OES) measurements (Table S1, Supplementary Material). The Ni/CdS/TiO2@MIL-101 material comprised 3.6 wt-% Ni, 18.8 wt-% CdS, and 26.0 wt-% TiO2. Diffuse reflectance ultraviolet-visible spectra in the range from 450 to 800 nm show an increased light absorption for TiO2@MIL-101 and CdS/TiO2@MIL-101 in comparison to MIL-101 (Fig. S5, Supplementary Material). The modification with CdS resulted in a distinctive edge for CdS/TiO2@MIL-101, which is comparable to the absorption of cubic CdS. We investigated the size and shape of the Ni/CdS/TiO2@MIL-101 crystallites by transmission electron microscopy (TEM) and high-angle annular dark-field scanning TEM (HAADF-STEM) measurements (Fig. 1). The typical octahedral shape and the homogeneous size distribution of the MIL-101 crystallites was also observed for Ni/CdS/TiO2@MIL-101. Energy dispersed X-ray maps demonstrate the uniform arrangement of TiO2, CdS, and Ni around single MIL-101 crystallites. The interface between the crystalline semiconductors CdS and TiO2 was verified by assigning characteristic lattice planes of adjacent semiconductor particles. Nickel was located on CdS and TiO2 and showed a minimal tendency for agglomeration (Fig. S5, Supplementary Material). We investigated the TiO2@MIL-101 and CdS@MIL-101 materials by TEM measurements to determine the size of the CdS and TiO2 crystallites formed on the surface of MIL-101 (Fig. S7, Supplementary Material). The average particle size was ~35 nm for TiO2 and 20 nm for CdS.
|
Time-resolved photoluminescence (TRPL) studies on photocatalytic systems allow a deeper understanding of photosynthetic electron dynamics between semiconductor materials and metal nanoparticles.[19] We confirmed the directed electron transfer process from excited CdS across the heterojunction interface to TiO2 and further to Ni nanoparticles by TRPL studies on MIL-101 supported catalysts. The photoluminescence lifetime of bare CdS@MIL-101 corresponds to the recombination rate of electrons and holes after optical excitation. The presence of TiO2 and Ni opens up additional relaxation channels of the excited CdS through the directed electron transfer induced by the potential gradient at the CdS/TiO2 and Ni/semiconductor interface. Thus, the photoluminescence lifetime of CdS, which can be calculated from the linear range of time-correlated single photon counting (TCSPC) traces in semi-log representation, is reduced (Fig. 2; Table S2, Supplementary Material). The charge carrier separation across three different catalyst components leads to the enhanced photocatalytic activity of Ni/CdS/TiO2@MIL-101. To extend the insight into the electron transfer between CdS and metallic species synchrotron-based X-ray absorption spectroscopy and photo-electrochemical characterisation could be combined.[20]

|
The photocatalytic liberation of hydrogen via the oxidation of alcohols leading to carbonyl compounds was performed under argon atmosphere at room temperature without additives or acceptor molecules. A 50 W blue LED (470 nm) was used as a visible light source (Fig. S9, Supplementary Material). The dehydrogenation of benzyl alcohol was investigated as a model reaction. Acetonitrile was identified as the most suitable solvent and a loading of 4 wt-% Ni showed the highest activity (Tables S3 and S4, Supplementary Material). We investigated the photocatalytic activity of CdS/TiO2@MIL-101 modified with the noble metals Pd, Pt, and Au as co-catalysts to ensure the superior performance of our Ni/CdS/TiO2@MIL-101 catalyst. The Ni-modified catalyst system showed the highest activity in alcohol splitting (Fig. S10a, Supplementary Material). In combination with CdS, the noble metal Pt is especially used as co-catalyst for hydrogen evolution reactions.[21]
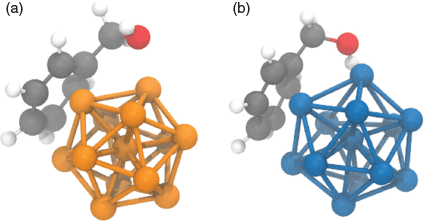
A detailed, microscopic understanding of the mechanism of such dehydrogenation reactions is a formidable task that is beyond the scope of this work. However, one straightforward first step towards understanding differences between Ni and noble metals as co-catalysts can be taken computationally. Of the three experimentally tested metals (Pd, Pt, and Au), we chose Pd as the paradigm noble metal catalyst for the comparison with Ni. Choosing Pd is motivated by the observation that similar metal particle geometries are stable for Ni and Pd, so binding properties can be readily compared. With the help of density functional theory (DFT) calculations, we checked whether there is a difference in the binding of benzyl alcohol to the metal particles using a molecular model consisting of a 13-atom metal particle and one benzyl alcohol molecule. We chose the 13-atom clusters, since they form stable structures[22] and can build an icosahedral geometry. The latter is one of the smallest possible faceted structures and, therefore, can be interpreted as a small model system for the binding to a faceted metal nanoparticle. We obtained the geometrical and electronic structure using the TURBOMOLE[23] code. Fig. 3 depicts the lowest energy, i.e. strongest bound, conformation in which the alcohol binds to the Ni13 and Pd13 cluster. Details of the computational procedures are reported in the Supplementary Material (see Theoretical Procedures). We paid particular attention to the fact that d-electron metals pose special challenges to many exchange-correlation approximations.[24] Reassuringly, our calculations with different density functionals consistently show that benzyl alcohol binds more strongly to Ni13 than to Pd13 by several hundred meV (Table S10, Supplementary Material). We therefore consider this trend as reliable. We see this calculation as one first, small step to corroborate the plausibility that Ni shows a different performance as a co-catalyst than Pd. Extensive further computational work, which is beyond the scope of the present manuscript, would be needed to reveal the detailed mechanism.
|
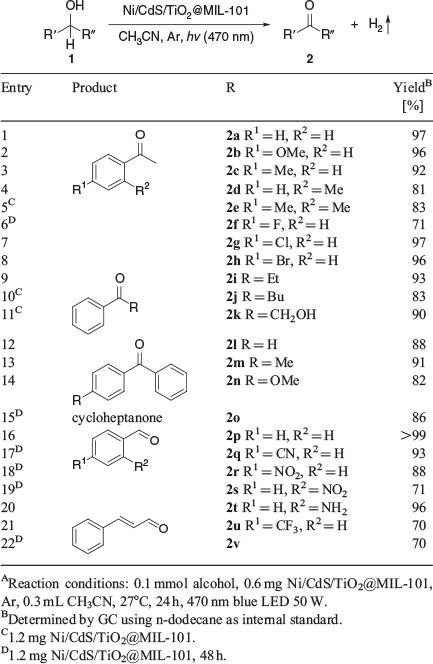
The beneficial contribution of the heterojunction between the semiconductors was demonstrated by the higher activity of CdS/TiO2@MIL-101 in comparison to CdS@MIL-101. We also demonstrated the favourable effect resulting from the support of Ni/CdS/TiO2 on MIL-101 as compared to pure Ni, CdS, TiO2, and Ni/CdS/TiO2. All materials without cadmium sulfide as the light-absorbing component showed no activity under visible light illumination (Table S5, Supplementary Material). The reusability of Ni/CdS/TiO2@MIL-101 was confirmed by performing ten successive runs without a remarkable loss of photocatalytic activity (Fig. S10b, Supplementary Material). The material was analysed by TEM and XRD measurements after the last run to demonstrate the structural integrity (Fig. S11, Supplementary Material). A light on/off experiment was performed to examine whether the dehydrogenation of benzyl alcohol is indeed a visible light-mediated reaction (Fig. S12, Supplementary Material). The amount of H2 released during the reaction was only increased under illumination. We further checked whether hydrogen is released in equimolar amounts during the photocatalytic reaction for several substrates using methane as an internal standard (Table S7, Supplementary Material). Performing the splitting of alcohols under argon atmosphere is crucial for high yields and the generation of molecular hydrogen (Table S7, Supplementary Material). Our Ni/CdS/TiO2@MIL-101 photocatalyst can split a broad range of alcohols 1 forming the corresponding aldehydes, aryl-alkyl, diaryl, and dialkyl ketones 2 (Table 1). A variety of functional groups was well tolerated, including halogens, methoxy, hydroxy, trifluoromethyl, and amino groups. In addition, hydrogenation-sensitive functionalities, such as nitrile and nitro groups, and C=C bonds can be tolerated selectively. We converted a total of 38 alcohols into the respective carbonyl compounds in good to excellent yields (Table 1; Table S6, Supplementary Material).
|
Finally, we were interested in photocatalytic C–N multiple bond formation reactions (Table 2). The reaction of benzyl alcohol and aniline was chosen to optimise the reaction conditions (Table S8, Supplementary Material). Imines 5 were obtained in good yields under very mild conditions at room temperature. A notable tolerance for functional group was again observed without the use of additives or sacrificial electron donors. Methyl, methoxy, halide, and hydroxy-substituted benzyl alcohols were well tolerated. In addition, we varied the amine component and found the tolerance for methyl, halogen, and methoxy substituents. We analysed the gas atmosphere to quantify the amount of hydrogen released for selected examples (Table S9, Supplementary Material). Again, no molecular hydrogen and a decreased yield of imine were observed when conducting the C–N bond formation without argon atmosphere. Amines formed via hydrogenation of corresponding imines by the hydrogen liberated were not detected.
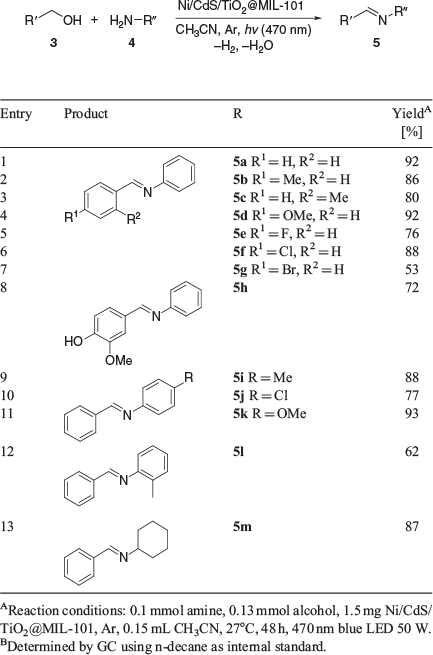
|
Conclusion
In conclusion, we introduced photocatalytic C–N multiple bond formation under visible light illumination by coupling activated carbonyl compounds with amines. The reusable, noble metal-free photocatalyst Ni/CdS/TiO2@MIL-101 permits quantitative hydrogen generation from alcohols (alcohol splitting) under very mild conditions, inert gas atmosphere, and without the requirement of sacrificial electron donors. The photocatalyst is composed of a CdS/TiO2 heterojunction with co-catalytic Ni nanoparticles assembled on the MOF MIL-101 as a colloidal porous support. We observed a broad substrate scope with very good tolerance of functional groups and high selectivity. We believe that our reusable photocatalyst represents a valuable platform for a variety of photocatalytic C–N or C–C bond formation reactions which involve the dehydrogenation of alcohols and condensation reaction(s) as key steps.
Supplementary Material
Details of the theoretical procedures, general methods, material synthesis and characterisation, and the general procedure for photocatalytic experiments and screening reactions are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interests.
Acknowledgements
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG, SFB 840(B1)). Note that parts of this work were included in the PhD thesis of Dominic Tilgner, Bayreuth 2017. The authors also acknowledge the support of the DAAD, Colloid/Polymer Network, the help of Florian Puchtler (XRD), Dr Jürgen Seidel (XPS), Prof. Dr Lothar Kador (FLIM) and Eduard Arzt for his support through INM. The photoluminescence lifetime measurements were performed in the keylab Electron and Optical Microscopy of the Bayreuth Polymer Institute (BPI).
References
[1] C. Gunanathan, D. Milstein, Science 2013, 341,| Crossref | GoogleScholarGoogle Scholar | 23869021PubMed |
[2] T. P. Vispute, H. Zhang, A. Sanna, R. Xiao, G. W. Huber, Science 2010, 330, 1222.
| Crossref | GoogleScholarGoogle Scholar | 21109668PubMed |
[3] C. O. Tuck, E. Perez, I. T. Horvath, R. A. Sheldon, M. Poliakoff, Science 2012, 337, 695.
| Crossref | GoogleScholarGoogle Scholar | 22879509PubMed |
[4] S. Michlik, R. Kempe, Nat. Chem. 2013, 5, 140.
| Crossref | GoogleScholarGoogle Scholar | 23344449PubMed |
[5] (a) B. Gnanaprakasam, J. Zhang, D. Milstein, Angew. Chem. Int. Ed. 2010, 49, 1468. [Angew. Chem. 2010, 122, 1510].
| Crossref | GoogleScholarGoogle Scholar |
(b) A. Mukherjee, A. Nerush, G. Leitus, L. J. W. Shimon, Y. Ben David, N. A. Espinosa Jalapa, D. Milstein, J. Am. Chem. Soc. 2016, 138, 4298.
| Crossref | GoogleScholarGoogle Scholar |
[6] (a) Pyrrole synthesis: D. Forberg, J. Obenauf, M. Friedrich, S. M. Hühne, W. Mader, G. Motz, R. Kempe, Catal. Sci. Technol. 2014, 4, 4188.
| Crossref | GoogleScholarGoogle Scholar |
(b) F. Kallmeier, B. Dudziec, T. Irrgang, R. Kempe, Angew. Chem. Int. Ed. 2017, 56, 7261.[Angew. Chem. 2017, 129, 7367].
| Crossref | GoogleScholarGoogle Scholar |
(c) For selected work of others see: D. Srimani, Y. Ben-David, D. Milstein, Angew. Chem. Int. Ed. 2013, 52, 4012.[Angew. Chem. 2013, 125, 4104].
| Crossref | GoogleScholarGoogle Scholar |
(d) M. Zhang, H. Neumann, M. Beller, Angew. Chem. Int. Ed. 2013, 52, 597.[Angew. Chem. 2013, 125, 625].
| Crossref | GoogleScholarGoogle Scholar |
(e) M. Zhang, X. Fang, H. Neumann, M. Beller, J. Am. Chem. Soc. 2013, 135, 11384.
| Crossref | GoogleScholarGoogle Scholar |
[7] (a) Pyridine synthesis: S. Michlik, R. Kempe, Angew. Chem. Int. Ed. 2013, 52, 6326.[Angew. Chem. 2013, 125, 6450].
| Crossref | GoogleScholarGoogle Scholar |
(b) T. Hille, T. Irrgang, R. Kempe, Angew. Chem. Int. Ed. 2017, 56, 371.[Angew. Chem. 2017, 129, 377].
| Crossref | GoogleScholarGoogle Scholar |
(c) For selected work of others see: D. Srimani, Y. Ben-David, D. Milstein, Chem. Commun. 2013, 49, 6632.
| Crossref | GoogleScholarGoogle Scholar |
[8] (a) Pyrimidine synthesis: N. Deibl, K. Ament, R. Kempe, J. Am. Chem. Soc. 2015, 137, 12804.
| Crossref | GoogleScholarGoogle Scholar | 26414993PubMed |
(b) N. Deibl, R. Kempe, Angew. Chem. Int. Ed. 2017, 56, 1663.[Angew. Chem. 2017, 129, 1685].
| Crossref | GoogleScholarGoogle Scholar |
(c) For selected work of others see: M. Mastalir, M. Glatz, E. Pittenauer, G. Allmaier, K. Kirchner, J. Am. Chem. Soc. 2016, 138, 15543.
| Crossref | GoogleScholarGoogle Scholar |
[9] (a) D. Forberg, T. Schwob, M. Zaheer, M. Friedrich, N. Miyajima, R. Kempe, Nat. Commun. 2016, 7, 13201.
| Crossref | GoogleScholarGoogle Scholar | 27762267PubMed |
(b) D. Forberg, T. Schwob, R. Kempe, Nat. Commun. 2018, 9, 1751.
| Crossref | GoogleScholarGoogle Scholar |
[10] (a) G. Zhang, S. K. Hanson, Org. Lett. 2013, 15, 650.
| Crossref | GoogleScholarGoogle Scholar | 23311959PubMed |
(b) J. Bain, P. Cho, A. Voutchkova-Kostal, Green Chem. 2015, 17, 2271.
| Crossref | GoogleScholarGoogle Scholar |
(c) P. Daw, S. Chakraborty, J. A. Garg, Y. Ben-David, D. Milstein, Angew. Chem. Int. Ed. 2016, 55, 14373.[Angew. Chem. 2016, 128, 14585].
| Crossref | GoogleScholarGoogle Scholar |
(d) M. Mastalir, M. Glatz, N. Gorgas, B. Stöger, E. Pittenauer, G. Allmaier, L. F. Veiros, K. Kirchner, Chem. – Eur. J. 2016, 22, 12316.
| Crossref | GoogleScholarGoogle Scholar |
(e) S. Parua, S. Das, R. Sikari, S. Sinha, N. D. Paul, J. Org. Chem. 2017, 82, 7165.
| Crossref | GoogleScholarGoogle Scholar |
[11] G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé, I. Margiolaki, Science 2005, 309, 2040.
| Crossref | GoogleScholarGoogle Scholar | 16179475PubMed |
[12] (a) For examples of a TiO2/MIL-101 core-shell material see: D. Tilgner, R. Kempe, Chem. – Eur. J. 2017, 23, 3184.
| Crossref | GoogleScholarGoogle Scholar | 27992084PubMed |
(b) D. Tilgner, M. Friedrich, A. Verch, N. de Jonge, R. Kempe, ChemPhotoChem 2018, 2, 349.
| Crossref | GoogleScholarGoogle Scholar |
[13] (a) Y. Shiraishi, Y. Sugano, S. Tanaka, T. Hirai, Angew. Chem. Int. Ed. 2010, 49, 1656.
| Crossref | GoogleScholarGoogle Scholar |
(b) T. P. A. Ruberu, N. C. Nelson, I. I. Slowing, J. Vela, J. Phys. Chem. Lett. 2012, 3, 2798.
| Crossref | GoogleScholarGoogle Scholar |
(c) T. Mitkina, C. Stanglmair, W. Setzer, M. Gruber, H. Kisch, B. Konig, Org. Biomol. Chem. 2012, 10, 3556.
| Crossref | GoogleScholarGoogle Scholar |
(d) Z. Chai, T.-T. Zeng, Q. Li, L.-Q. Lu, W.-J. Xiao, D. Xu, J. Am. Chem. Soc. 2016, 138, 10128.
| Crossref | GoogleScholarGoogle Scholar |
(e) D. Jiang, X. Chen, Z. Zhang, L. Zhang, Y. Wang, Z. Sun, R. M. Irfan, P. Du, J. Catal. 2018, 357, 147.
| Crossref | GoogleScholarGoogle Scholar |
[14] (a) W. Zhai, S. Xue, A. Zhu, Y. Luo, Y. Tian, ChemCatChem 2011, 3, 127.
| Crossref | GoogleScholarGoogle Scholar |
(b) S. Higashimoto, Y. Tanaka, R. Ishikawa, S. Hasegawa, M. Azuma, H. Ohue, Y. Sakata, Catal. Sci. Technol. 2013, 3, 400.
| Crossref | GoogleScholarGoogle Scholar |
(c) Z. Liu, J. Caner, A. Kudo, H. Naka, S. Saito, Chem. – Eur. J. 2013, 19, 9452.
| Crossref | GoogleScholarGoogle Scholar |
(d) A. Tanaka, S. Sakaguchi, K. Hashimoto, H. Kominami, ACS Catal. 2013, 3, 79.
| Crossref | GoogleScholarGoogle Scholar |
(e) H. Kasap, C. A. Caputo, B. C. M. Martindale, R. Godin, V. W.-h. Lau, B. V. Lotsch, J. R. Durrant, E. Reisner, J. Am. Chem. Soc. 2016, 138, 9183.
| Crossref | GoogleScholarGoogle Scholar |
(f) L. M. Zhao, Q. Y. Meng, X. B. Fan, C. Ye, X. B. Li, B. Chen, V. Ramamurthy, C. H. Tung, L. Z. Wu, Angew. Chem. Int. Ed. 2017, 56, 3020.[Angew. Chem. 2017, 129, 3066].
| Crossref | GoogleScholarGoogle Scholar |
(g) G. Han, Y.-H. Jin, R. A. Burgess, N. E. Dickenson, X.-M. Cao, Y. Sun, J. Am. Chem. Soc. 2017, 139, 15584.
| Crossref | GoogleScholarGoogle Scholar |
[15] J. Hermannsdörfer, M. Friedrich, R. Kempe, Chem. – Eur. J. 2013, 19, 13652.
| Crossref | GoogleScholarGoogle Scholar | 24092537PubMed |
[16] (a) J. He, Z. Yan, J. Wang, J. Xie, L. Jiang, Y. Shi, F. Yuan, F. Yu, Y. Sun, Chem. Commun. 2013, 49, 6761.
| Crossref | GoogleScholarGoogle Scholar |
(b) Y. Wang, Y. Zhang, Z. Jiang, G. Jiang, Z. Zhao, Q. Wu, Y. Liu, Q. Xu, A. Duan, C. Xu, Appl. Catal. B 2016, 185, 307.
| Crossref | GoogleScholarGoogle Scholar |
(c) Z. Jiang, J. Liu, M. Gao, X. Fan, L. Zhang, J. Zhang, Adv. Mater. 2017, 29,
| Crossref | GoogleScholarGoogle Scholar |
[17] (a) H. Gerischer, M. Lübke, J. Electroanal. Chem. 1986, 204, 225.
| Crossref | GoogleScholarGoogle Scholar |
(b) L. Spanhel, H. Weller, A. Henglein, J. Am. Chem. Soc. 1987, 109, 6632.
| Crossref | GoogleScholarGoogle Scholar |
(c) K. R. Gopidas, M. Bohorquez, P. V. Kamat, J. Phys. Chem. 1990, 94, 6435.
| Crossref | GoogleScholarGoogle Scholar |
(d) R. Vogel, K. Pohl, H. Weller, Chem. Phys. Lett. 1990, 174, 241.
| Crossref | GoogleScholarGoogle Scholar |
(e) S. Kohtani, A. Kudo, T. Sakata, Chem. Phys. Lett. 1993, 206, 166.
| Crossref | GoogleScholarGoogle Scholar |
(f) R. Vogel, P. Hoyer, H. Weller, J. Phys. Chem. 1994, 98, 3183.
| Crossref | GoogleScholarGoogle Scholar |
(g) J. E. Evans, K. W. Springer, J. Z. Zhang, J. Chem. Phys. 1994, 101, 6222.
| Crossref | GoogleScholarGoogle Scholar |
(h) D. Baker, P. V. Kamat, Adv. Funct. Mater. 2009, 19, 805.
| Crossref | GoogleScholarGoogle Scholar |
[18] (a) T. Simon, N. Bouchonville, M. J. Berr, A. Vaneski, A. Adrović, D. Volbers, R. Wyrwich, M. Döblinger, A. S. Susha, A. L. Rogach, F. Jäckel, J. K. Stolarczyk, J. Feldmann, Nat. Mater. 2014, 13, 1013.
| Crossref | GoogleScholarGoogle Scholar | 25087066PubMed |
(b) Y. Xu, R. Xu, Appl. Surf. Sci. 2015, 351, 779.
| Crossref | GoogleScholarGoogle Scholar |
[19] (a) R. Shi, Y. Cao, Y. Bao, Y. Zhao, G. I. N. Waterhouse, Z. Fang, L.-Z. Wu, C.-H. Tung, Y. Yin, T. Zhang, Adv. Mater. 2017, 29,
| Crossref | GoogleScholarGoogle Scholar | 28497896PubMed |
(b) H. Zhao, X. Ding, B. Zhang, Y. Li, C. Wang, Sci. Bull. 2017, 62, 602.
| Crossref | GoogleScholarGoogle Scholar |
[20] Q. He, Y. Wang, Y. Zhang, H. Jiang, H. Liu, X. Zheng, S. Chen, X. Wu, L. Song, Sol. RRL 2018,
[21] (a) J. He, Z. Yan, J. Wang, J. Xie, L. Jiang, Y. Shi, F. Yuan, F. Yu, Y. Sun, Chem. Commun. 2013, 49, 6761.
| Crossref | GoogleScholarGoogle Scholar |
(b) L. Shang, B. Tong, H. Yu, G. I. N. Waterhouse, C. Zhou, Y. Zhao, M. Tahir, L.-Z. Wu, C.-H. Tung, T. Zhang, Adv. Energy Mater. 2016, 6,
| Crossref | GoogleScholarGoogle Scholar |
[22] (a) A. Langenberg, K. Hirsch, A. Ławicki, V. Zamudio-Bayer, M. Niemeyer, P. Chmiela, B. Langbehn, A. Terasaki, B. v. Issendorff, J. T. Lau, Phys. Rev. B Condens. Matter Mater. Phys. 2014, 90,
| Crossref | GoogleScholarGoogle Scholar |
(b) J. Meyer, M. Tomber, T. van Wüllen, G. Niedner-Schattenburg, S. Peredkob, W. Eberhardt, M. Neeb, S. Palutke, M. Martins, W. Wurth, J. Chem. Phys. 2015, 143,
| Crossref | GoogleScholarGoogle Scholar |
(c) M. B. Knickelbein, J. Chem. Phys. 2002, 116, 9703.
| Crossref | GoogleScholarGoogle Scholar |
(d) S. E. Apsel, J. W. Emmert, J. Deng, L. A. Bloomfield, Phys. Rev. Lett. 1996, 76, 1441.
| Crossref | GoogleScholarGoogle Scholar |
(e) A. J. Cox, J. G. Louderback, S. E. Apsel, L. A. Bloomfield, Phys. Rev. B Condens. Matter 1994, 49, 12295.
| Crossref | GoogleScholarGoogle Scholar |
[23] TURBMOLE V7.3 2018 (University of Karlsruhe and Forschungszentrum Karlsruhe GmbH: Karlsruhe, Germany).
[24] (a) C. J. Cramer, D. G. Truhlar, Phys. Chem. Chem. Phys. 2009, 11, 10757.
| Crossref | GoogleScholarGoogle Scholar | 19924312PubMed |
(b) L. Leppert, R. Kempe, S. Kümmel, Phys. Chem. Chem. Phys. 2015, 17, 26140.
| Crossref | GoogleScholarGoogle Scholar |
(c) T. Schmidt, S. Kümmel, Computation 2016, 4, 33.
| Crossref | GoogleScholarGoogle Scholar |
* This paper is dedicated to Richard Robson, an outstanding pioneer in the field of porous coordination polymers and metal-organic frameworks.