

Corrigendum to: Valencene derived from essential oils of Psidium guajava L. as multi-target neurodegenerative inhibitor: a computational study
Ram Lal Swagat Shrestha A , Sujan Dhital A , Nirmal Parajuli A , Prabhat Neupane A , Manila Poudel B , Timila Shrestha A , Samjhana Bharati A , Binita Maharjan A , Bishnu Prasad Marasini C * and Jhashanath Adhikari Subin D *
A , Nirmal Parajuli A , Prabhat Neupane A , Manila Poudel B , Timila Shrestha A , Samjhana Bharati A , Binita Maharjan A , Bishnu Prasad Marasini C * and Jhashanath Adhikari Subin D *
A
B
C
D
Handling Editor: Amir Karton
Abstract
The prevalence of neurodegenerative disorders such as Alzheimer’s, Parkinson’s and Huntington’s has been gradually increasing in recent times. These diseases could respectively be treated through the inhibition of acetylcholinesterase (AChE), monoamine oxidase B (MAOB) and huntingtin (HTT) proteins. This study aims to identify the compounds present in the oil of Psidium guajava L. using GC-MS analysis and the potent neurodegenerative inhibitors through computational methods. The results revealed the presence of 42 different phytocompounds, and molecular docking calculations demonstrated firm binding of Valencene with the receptor proteins AChE, MAOB and HTT, with respective binding affinities of −9.246, −9.794 and −9.541 kcal mol–1. These values are better than that of reference drugs Selegiline, Rivastigmine and Ciproxifan with respective binding affinities of −8.187, −8.883 and −10.03 kcal mol–1. All three complexes, MAOB–Valencene, AChE–Valencene and HTT–Valencene demonstrated good geometrical stability, showing smooth RMSD curves and ligand RMSD of ~3.5, ~3.0 and ~6.0 Å respectively, from 100-ns molecular dynamics simulation. The thermodynamic stability assessed through the MMPBSA method in terms of binding free energy changes revealed sustained spontaneity and feasibility of the adduct formations. The pharmacokinetic prediction supported the drug-like properties of Valencene. The comparative geometric stability and better thermodynamic stability of the Valencene complexes, compared to the reference drugs targeting key molecular sites in neurodegenerative diseases (MAOB, AChE and HTT), suggest that it could be a hit candidate. Therefore, validation of Valencene through in vivo and in vitro experiments is recommended, as it could be a potential remedy for neurological disorders.
Keywords: ADMET predictions, binding free energy changes, essential oil, molecular docking, molecular dynamics simulations, neurodegenerative disorders, Psidium guajava.
Notes on this corrigendum
This corrigendum corrects Australian Journal of Chemistry 78, CH24117. doi:10.1071/CH24117
The authors of the above-mentioned paper regret to inform readers that the wrong version of this paper was published. When the manuscript was at the revision stage in peer review, both the original submission and the revised version were uploaded, but it was the former version that was typeset and then published. That incorrect version was missed when the paper was at the proof stage.
As there are substantial differences between these versions, this corrigendum includes the complete paper with the differences highlighted by bold formatting.
We apologise for that error and any confusion this may have caused.
Introduction
Neurodegeneration, characterised by the gradual dysfunction and deterioration of neurons and axons within the central nervous system, stands as the predominant pathological hallmark of both acute and chronic neurodegenerative disorders like Alzheimer’s disease (AD), Parkinson’s disease (PD) and Huntington’s disease (HD).1 The occurrence of neurodegenerative diseases is anticipated to rise with increasing lifespan in most countries.2 Currently, ~50 million individuals are affected by dementia, a number projected to surge to 130 million by 2050.3 AD is the most prevalent form of neurodegenerative disorder resulting in a gradual decline in cognitive function and is becoming one of the most costly, deadliest and burdensome illnesses.4 Similarly, PD stands as the second most prevalent neurodegenerative disorder, affecting over 6 million people worldwide.5 Parkinson’s syndrome is distinguished by the loss of dopaminergic cells and observable irregularities in the spontaneous activity and sensorimotor responses of neurons.6 Likewise, another inherited neurodegenerative disease, HD, is caused by the repetition of CAG sequences (36 repeats or more) in the initial exon of the huntingtin (HTT) protein. This results in an anomalous form of the HTT protein that clumps together to form aggregates,7 characterised by uncontrollable, exaggerated motor movements, alongside emotional and cognitive impairments.8 AD could be treated by inhibiting acetylcholinesterase (AChE), a crucial enzyme responsible for the degradation of acetylcholine, thereby enhancing the effectiveness of brain signaling. AChE inhibitors prevent the breakdown of acetylcholine, consequently fostering enhancements in cholinergic neurotransmission.9 Analogously, inhibition of Monoamine Oxidase B (MAOB) could lead to effective treatment of PD.10 In patients affected with PD, high levels of MAOB in the brain and blood platelets have been observed, and blocking this enzyme could potentially provide a pathway for curing the condition.11 HD can be addressed by inhibiting the aggregation of mutant HTT by targeting the potential sites that facilitate protein aggregation, thus potentially slowing down the disease’s progression.12
Psidium guajava L. (P. guajava) belongs to the Myrtaceae family and is a commercial crop in numerous subtropical and tropical regions across the globe.13 Owing to the presence of a large number of phytocompounds in the leaves of the guava plant, it has been used for various health benefits such as anti-diabetic, antioxidant, anticancer, antimicrobial and hepatoprotection activities since ancient times.14 The leaves have also been used to treat neurodegenerative conditions like AD.15,16 Numerous studies have been performed using various computational approaches as they are known for their cost effectiveness and efficiency in the drug discovery process.17 The binding affinity and putative binding mode between the receptors and ligands can be identified using computational techniques.18 The aim of this research was to identify the phytocompounds present in the oil of guava leaves using GC-MS analysis and to study their ability to manage neurodegenerative disorders computationally.
Results and discussion
GC-MS analysis
The analysis of the GC-MS chromatogram revealed 42 peaks in the oil of P. guajava, as shown in the Supplementary material (Fig. S1). It confirmed the presence of 42 compounds, with limonene being the most prevalent one, constituting 30.15% of the area percentage. The mass spectra of each phytochemical, along with its structure, are depicted in the Supplementary Fig. S2 and S3. The name, peak number, molecular weight, area percentage, molecular formula and retention time of the obtained compounds are shown in Table 1.
| Peak number | Name of compounds | Molecular formula | Molecular weight (g mol–1) | Retention time (min) | Area (%) | |
|---|---|---|---|---|---|---|
| 1 | α-Pinene | C10H16 | 136.23 | 9.435 | 0.51 | |
| 2 | Myrcene | C10H16 | 136.23 | 11.749 | 0.68 | |
| 3 | 1,4,4-Trimethyl-3,5-dimethylidenecyclopentene | C10H14 | 134.22 | 13.262 | 0.26 | |
| 4 | Limonene | C10H16 | 136.23 | 13.488 | 30.15 | |
| 5 | Eucalyptol | C10H18O | 154.25 | 13.606 | 1.19 | |
| 6 | (E)-β-ocimene | C10H16 | 136.23 | 13.856 | 0.36 | |
| 7 | Lavandulyl 2-methylbutyrate | C15H26O2 | 238.37 | 29.311 | 0.22 | |
| 8 | α-Copaene | C15H24 | 204.35 | 29.935 | 4.54 | |
| 9 | (Z)-caryophyllene | C15H24 | 204.35 | 31.584 | 1.08 | |
| 10 | (E)-Caryophyllene | C15H24 | 204.35 | 31.948 | 16.63 | |
| 11 | Valencene | C15H24 | 204.35 | 33.231 | 0.49 | |
| 12 | α-Humulene | C15H24 | 204.35 | 33.425 | 2.88 | |
| 13 | 9-epi-(E)-caryophyllene | C15H24 | 204.35 | 33.753 | 0.47 | |
| 14 | γ-Muurolene | C15H24 | 204.35 | 34.385 | 0.36 | |
| 15 | Trans-bergamotol | C15H24O | 220.35 | 34.532 | 0.50 | |
| 16 | β-Selinene | C15H24 | 204.35 | 34.857 | 0.29 | |
| 17 | α-amorphene | C15H24 | 204.35 | 35.407 | 0.77 | |
| 18 | β-Bisabolene | C15H24 | 204.35 | 35.678 | 0.39 | |
| 19 | Spathulenol | C15H24O | 220.35 | 36.074 | 5.28 | |
| 20 | Trans-calamenene | C15H22 | 202.33 | 36.381 | 3.59 | |
| 21 | Trans-cadina-1,4-diene | C15H24 | 204.35 | 36.762 | 1.10 | |
| 22 | α-Dehydro-ar-himachalene | C15H20 | 200.32 | 37.061 | 0.22 | |
| 23 | α-Calacorene | C15H20 | 200.32 | 37.227 | 0.51 | |
| 24 | 14-Hydroxy-4,5-dihydro-β-caryophyllene | C15H26O | 222.37 | 37.649 | 0.79 | |
| 25 | Z-Nerolidyl acetate | C17H28O2 | 264.4 | 37.930 | 7.47 | |
| 26 | Caryolan-8-ol | C15H26O | 222.37 | 38.414 | 0.33 | |
| 27 | Aromadendrene | C15H24 | 204.35 | 39.784 | 2.59 | |
| 28 | Copaene | C15H24 | 204.35 | 40.205 | 0.23 | |
| 29 | Cis-calamenene | C15H22 | 202.33 | 40.300 | 0.25 | |
| 30 | 14-Hydroxy-9-epi-(E)-caryophyllene | C15H24O | 220.35 | 40.540 | 0.55 | |
| 31 | α-Cubebene | C15H24 | 204.35 | 40.726 | 3.10 | |
| 32 | Allo-aromadendrene epoxide | C15H24O | 220.35 | 40.840 | 3.32 | |
| 33 | Cis-p-Mentha-1(7),8-dien-2-ol | C10H16O | 152.23 | 40.920 | 0.97 | |
| 34 | Caryophylla-4(12),8(13)-dien-5-ol | C15H24O | 220.35 | 41.076 | 0.47 | |
| 35 | Guaiac acetate | C17H28O2 | 264.4 | 41.200 | 1.05 | |
| 36 | Epicubenol | C15H26O | 222.37 | 41.278 | 1.67 | |
| 37 | α-Muurolol | C15H26O | 222.37 | 41.425 | 1.67 | |
| 38 | δ-Amorphene | C15H24 | 204.35 | 41.755 | 0.94 | |
| 39 | α-Himachalene | C15H24 | 204.35 | 42.340 | 0.43 | |
| 40 | 14-hydroxy-Caryophyllene | C15H24O | 220.35 | 42.416 | 0.90 | |
| 41 | (E)-β-farnesene | C15H26 | 206.37 | 44.176 | 0.56 | |
| 42 | Nootkatene | C15H22 | 202.33 | 50.363 | 0.27 |
Binding affinity from molecular docking calculations
The molecular docking calculation was used to assess the best binding pose of the ligands within the active site of the receptor protein, in terms of binding affinity.19 The results of molecular docking with the MAOB protein revealed β-bisabolene, nootkatene, allo-aromadendrene epoxide, α-copaene and Valencene as the top five best ligands, with binding affinities ranging from −9.993 to −9.794 kcal mol–1, signifying stronger interaction and binding with the receptor. The highest binding affinity was demonstrated by β-bisabolene with −9.993 kcal mol–1. The binding affinity of Valencene was −9.794 kcal mol–1, greater than that of the reference drug Selegiline (−8.817 kcal mol–1) with the target MAOB.
From the molecular docking calculation with the HTT protein, the best binding affinity was observed with β-bisabolene with a binding affinity of −9.776 kcal mol–1, followed by 9-epi-(E)-caryophyllene and allo-aromadendrene epoxide, with −9.691 and −9.666 kcal mol–1 respectively. Similarly, the binding affinity of −9.541 kcal mol–1 was observed for Valencene. The reference drug Ciproxifan scored binding affinity −10.034 kcal mol–1, which was found to be slightly higher than that of selected ligands in terms of affinity towards HTT protein.
In the case of the AChE protein, the outcome of molecular docking demonstrated the best binding affinity of −9.559 kcal mol–1 for guaiac acetate. The binding affinities of −9.526 and −9.305 kcal mol–1 were obtained for Z-nerolidyl acetate and β-bisabolene respectively. Likewise, the binding affinity of −9.246 kcal mol–1 was shown by Valencene, which was found to be comparatively better than that of the reference drug, Rivastigmine (−8.883 kcal mol–1).
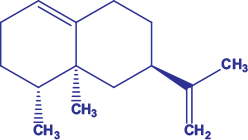
Among the studied compounds, the molecular docking of β-bisabolene, allo-aromadendrene epoxide, Valencene (Fig. 1) and nootkatene exhibited better binding affinities with all the neurodegenerative receptor proteins MAOB, huntingtin and acetylcholinesterase, as shown in Table 2 and Supplementary Table S1.
| Series number | Ligands | PubChem CID | Binding affinity (kcal mol–1) | |||
|---|---|---|---|---|---|---|
| MAOB | HTT | AChE | ||||
| 1 | β-Bisabolene | 10104370 | −9.993 | −9.776 | −9.305 | |
| 2 | Allo-aromadendrene epoxide | 91746712 | −9.803 | −9.666 | −9.247 | |
| 3 | Nootkatene | 25200342 | −9.831 | −9.554 | −9.187 | |
| 4 | Valencene | 9855795 | −9.794 | −9.541 | −9.246 | |
| 5 | α-Copaene | 70678558 | −9.802 | −9.515 | −9.089 | |
| 6 | E-Caryophyllene | 5281515 | −9.75 | −9.606 | −9.10 | |
| 7 | α-Humulene | 5281520 | −9.745 | −9.548 | −9.114 | |
| 8 | α-Himachalene | 520909 | −9.74 | −9.503 | −9.188 | |
| 9 | Cis-calamenene | 6429077 | −9.719 | −9.384 | −9.028 | |
| 10 | α-Dehydro-ar-himachalene | 15098633 | −9.705 | −9.419 | −9.138 | |
| 11 | Native | – | −10.551 | −9.785 | −11.183 | |
| 12 | Selegiline | 26757 | −8.817 | – | – | |
| 13 | Ciproxifan | 6422124 | – | −10.034 | – | |
| 14 | Rivastigmine | 77991 | – | – | −8.883 | |
Valencene is denoted in bold (and italic) for quick identification and comparison.
Protein–ligand interactions
The three-dimensional (3-D) and two-dimensional (2-D) representations of the interactions between the ligand and the amino acid residue of the proteins are shown in Fig. 2. It depicts the presence of only hydrophobic interactions, like pi–alkyl, alkyl, pi–sigma and van der Waals between the ligand and receptor proteins, as shown in Table 3. In the interaction of Valencene with the MAOB (4A79) protein, pi–alkyl interactions were observed with the amino acid residues PHE101, TRP117 and TYR324. Likewise, PRO102, LEU162, ILE197 and ILE 314 showed alkyl interaction with the ligand. In the Valencene–HTT (7F61) protein complex, the amino acid residues TYR67, TRP83, TYR88, TYR162, PHE166, TYR262, PHE266 and TRP270 exhibited pi–alkyl interactions with the ligand. The alkyl and pi–sigma interaction was observed with amino acid residues LEU84 and PHE266 respectively. For the hydrophobic interaction, pi–alkyl interactions were observed between the Valencene and the amino acid residues TRP83, TYR238, PHE329, TYR332 and HIS438 of the AChE (7E3H) protein. Several van der Waals interactions were observed between the ligand (Valencene) and different receptor proteins. The interaction between the Valencene molecule and the amino acid residues of the proteins depicted a strong hydrophobic interaction, inferring its potential to inhibit the normal functioning of acetylcholinesterase, monoamine oxidase B and huntingtin proteins.
3-D (left) and 2-D (right) representations of the docked ligand at the binding site of the (a) MAOB (4A79), (b) HTT (7F61) and (c) AChE (7E3H) proteins. [Readers should note that the layout of this figure has changed.]
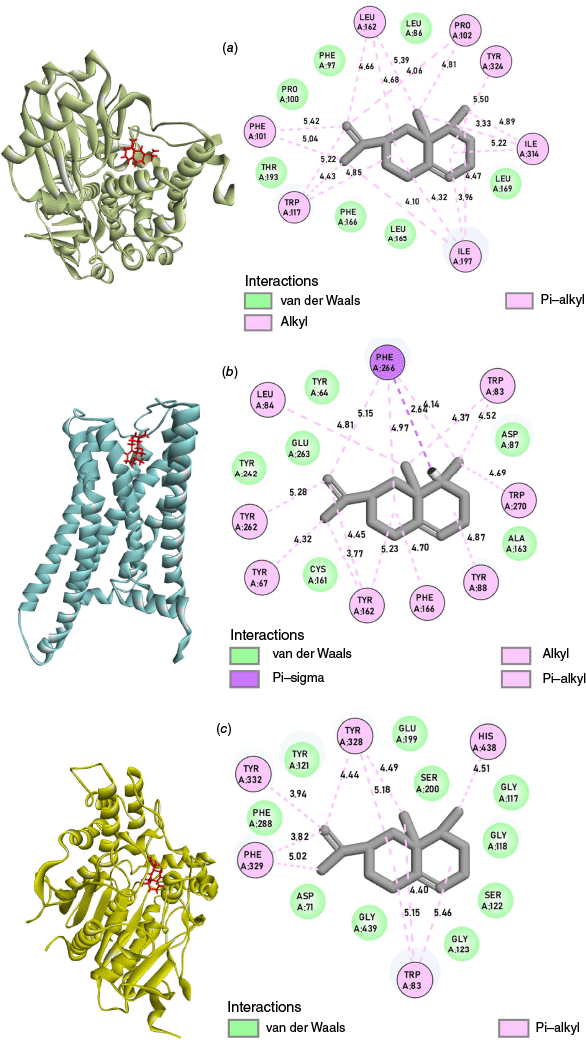
| Proteins | Types of interaction | Active site residues (Å) | |
|---|---|---|---|
| MAOB (4A79) | Alkyl | PRO102 (4.06, 4.81), LEU162 (4.66, 4.68, 5.39), ILE197 (3.96, 4.10, 4.32, 4.47), ILE314 (3.33, 4.89, 5.22) | |
| Pi–alkyl | PHE101 (5.04, 5.42), TRP117 (4.43, 4.85, 5.22), TYR324 (5.50) | ||
| Van der Waals | LEU86, PHE97, PRO100, LEU165, PHE166, LEU169, THR193 | ||
| HTT (7F61) | Alkyl | LEU84 (4.81) | |
| Pi–alkyl | TYR67 (4.32), TRP83 (4.37, 4.52), TYR88 (4.87), TYR162 (3.77, 4.45, 5.23), PHE166 (4.70), TYR262 (5.28), PHE266 (4.14, 4.97, 5.15), TRP270 (4.69) | ||
| Pi–sigma | PHE266 (2.64) | ||
| Van der Waals | TYR64, ASP87, CYS161, ALA163, TYR242, GLU263 | ||
| AChE (7E3H) | Pi–alkyl | TRP83 (4.40, 5.15, 5.46), TYR328 (4.44, 4.49, 5.18), PHE329 (3.82, 5.02), TYR332 (3.94), HIS438 (4.51) | |
| Van der Waals | ASP71, GLY117, GLY118, TYR121, SER122, GLY123, GLU199, SER200, PHE288, GLY439 |
Molecular dynamics simulation (MDS) analysis
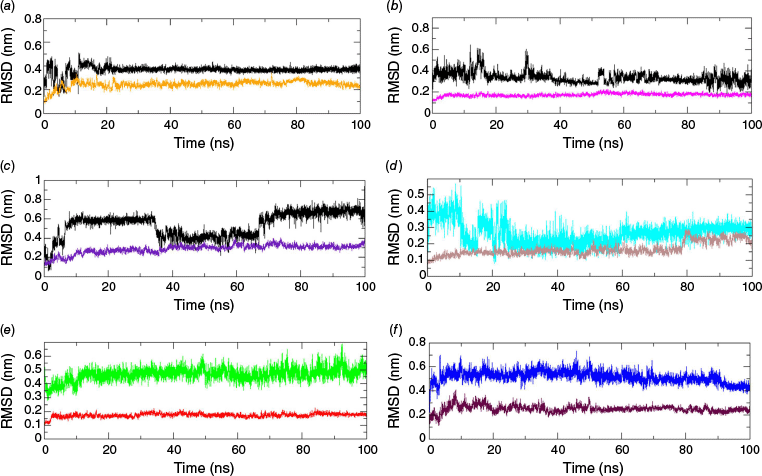
MD simulation of the top ligands from docking calculations and reference drugs complexed with different target proteins was performed to analyse the stability of the adduct formed. The geometrical stability of ligands in their adducts can be assessed through the root mean square deviation (RMSD) profiling obtained from the MDS trajectory.20 The RMSD of the protein backbone and ligand with respect to the protein backbone depicts the overall stability of the formed adducts as shown in Fig. 3.21 Among top-docked ligands, the molecular dynamics simulation of Valencene showed a good stability with proteins (PDB ID: 4A79, 7F61 and 7E3H), with RMSD below 6.5 Å. The RMSD plots of the other three ligands, with respect to the protein backbone, were found to be inconsistent throughout the production run and are presented in Supplementary Fig. S4 and S5. The RMSD trajectories of aromadendrin (Fig. S4) and bisabolene (Fig. S5) with different target proteins were found to be reasonably unstable with high RMSD values. The reasonably smooth RMSD curve of the ligand demonstrated the stability of the adduct during the production run in terms of the conservation of the docked pose and position. An exceptional stability was shown by Valencene with the MAOB protein, with a consistently smoother RMSD trajectory of ~4 Å despite some initial spikes (Fig. 3a). The ligand RMSD of ~3.0 Å, with slight fluctuation and spikes, was depicted by Valencene with the AChE protein, implying that the ligand remained docked at the same location throughout the production run (Fig. 3b). The Valencene exhibited moderate stability with the HTT protein, with a RMSD of ~6.0 Å, along with some fluctuations (Fig. 3c). Despite major fluctuations and spikes in the trajectory, the system attained equilibrium after 70 ns. Although the reference Selegiline was found to be stable after 30 ns with a RMSD of ~2.5 Å in complexation with MAOB protein (Fig. 3d), Valencene depicted a more stable trajectory at 4 Å with the same target. Ligand RMSD of another reference drug Rivastigmine complexed with AChE, showed a gradually increasing trajectory, initiating from 4 Å up to 100-ns production run (Fig. 3e), which was found to be comparable to the trajectory formed by ligand RMSD in the Valencene–AChE complex. Likewise, the Ligand RMSD trajectory in the HTT–Ciproxifan complex was found to be stable at 5 Å with few spikes at initiation and end of the production run (Fig. 3F). So, the ligand RMSDs of the Valencene with multiprotein clearly showed their comparable geometric stability as found in various reference drugs targeting three neurodegenerative proteins. A smooth curve with a RMSD of ~2.0 Å of the protein backbone inferred the stability of protein structure upon ligand binding. No significant structural changes were observed.
RMSD of Valencene, protein backbone and reference drugs (Selegiline, Rivastigmine and Ciproxifan) with respect to protein backbone in different adducts (a) MAOB (orange)–Valencene (black), (b) AChE (magenta)–Valencene (black), (c) HTT (indigo)–Valencene (black), (d) MAOB (brown)–Selegiline (cyan), (e) AChE (red)–Rivastigimine (green) and (f) HTT (maroon)–Ciproxifan (blue). [Readers should note that the layout of this figure has changed.]
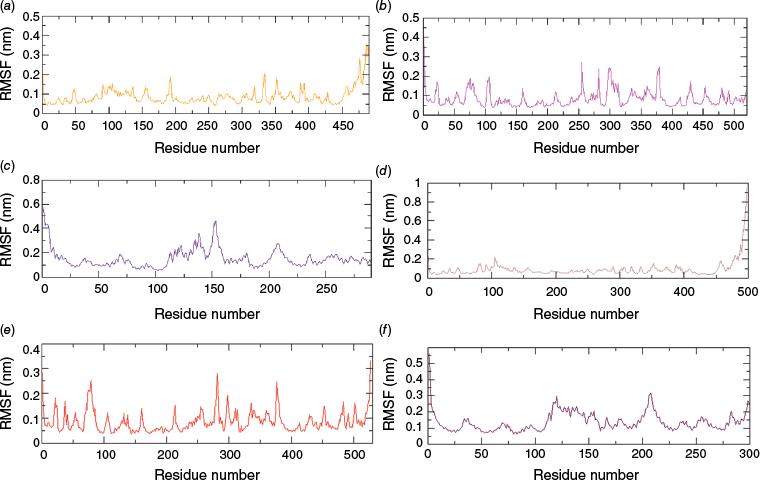
The root mean square fluctuation (RMSF) depicts the extent of fluctuation and flexibility of the amino acid residues of a protein over the 100-ns production run. Higher RMSF indicates greater flexibility during simulation, whereas lower RMSF indicates minimal changes, implying a tightly bound protein–ligand complex and enhanced system stability.22 The RMSF plot (Fig. 4a) shows that the fluctuation of α-carbon atoms of the MAOB protein was less than 2.0 Å for almost all amino acid residues except for the terminal part. Similarly, the RMSF of the amino acid residue of the AChE and HTT proteins were less than 3.0 Å, with an exception at ~140 and 155 amino acid residue numbers of the 7F61 protein. The RMSF trajectory of AChE and HTT proteins with Valencene and their respective reference drugs (Rivastigmine and Ciproxifan) are observed to be comparatively similar (Fig. 4). Surprisingly, RMS fluctuation of protein α-carbon atoms in the MAOB–Valencene complex was observed to be comparatively higher than found in the MAOB–Selegiline complex (Fig. 3d).
RMSF plots of protein backbones in complexes (a) MAOB–Valencene (orange), (b) AChE–Valencene (magenta), (c) HTT–Valencene (indigo), (d) MAOB–Selegiline (Brown), (e) AChE–Rivastigimine (red) and (f) HTT–Ciproxifan (maroon). [Readers should note that the layout of this figure has changed.]
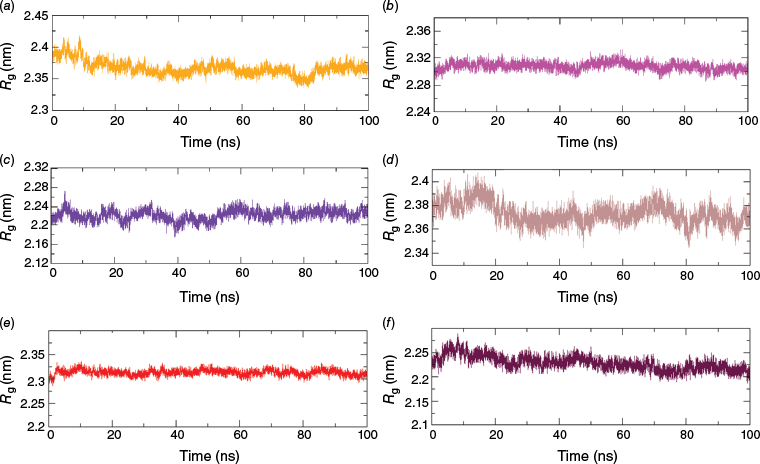
The radius of gyration (Rg) obtained through MDS reflects the compactness of protein–ligand complexes, with a smaller Rg suggesting a more compact structure.23 From the calculation of Rg, a steady and smooth trajectory at ~23 ± 0.7 Å indicated no significant expansion or shrinkage of the proteins, implying the stability of all three proteins (4A79, 7E3H and 7F61) upon the binding of the ligand as evident from Fig. 5. The Rg of protein obtained for AChE (Fig. 5b) and HTT (Fig. 5e) docked with reference drugs were found to be similar to those found for the docked complex with Valencene at ~23 and ~22 Å respectively. The Rg plot of the MAOB protein docked with the drug Selegiline was found to have various spikes throughout the MDS run (Fig. 5d), but the MAOB complexed with Valencene was comparatively smooth. Such an undatable Rg trajectory of the protein with Selegiline indicates a more stable complex formation of MAOB–Valencene than the Selegiline–MAOB complex.
Rg of protein in complexes (a) MAOB–Valencene (orange), (b) AChE–Valencene (magenta), (c) HTT–Valencene (indigo), (d) MAOB–Selegiline (Brown), (e) AChE–Rivastigimine (red) and (f) HTT–Ciproxifan (maroon). [Readers should note that the layout of this figure has changed.]
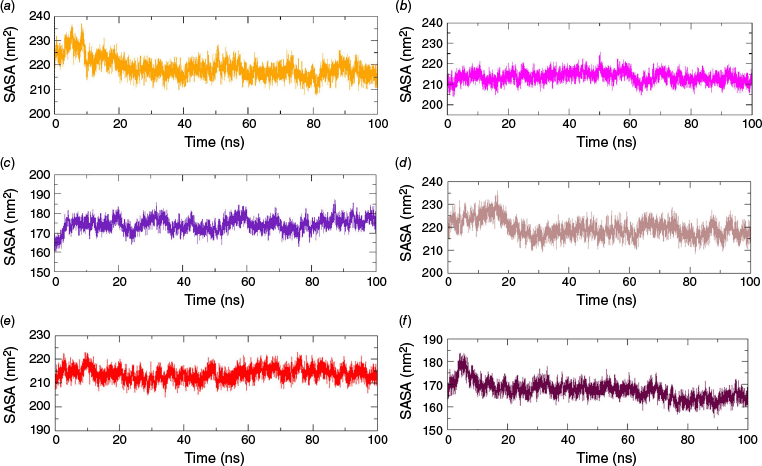
The solvent-accessible surface areas (SASAs) refer to the area of a protein’s surface that interacts with its solvent molecules.24 A reasonably smooth trajectory with SASAs of 216 ± 4 nm2 was observed with MAOB–Valencene and AChE–Valencene complexes. Likewise, a smooth SASA curve with a value of ~175 nm2 was obtained for the HTT–Valencene complex (Fig. 6). The SASAs remained almost stable throughout the production run without any significant bumps and spikes, indicating no change in the wettable area of the protein upon ligand binding over time in the Valencene complexes. Though few initial bumps were noticed in MAOB (Fig. 6d) and HTT (Fig. 6f) complexed with the reference drug, the SASA of all the reference drugs was observed to be similar to that of the complex formed by Valencene with different proteins, which is presented in Fig. 6. The combined analysis of all the geometric parameters, RMSD, SASA, RMSF and Rg demonstrated the good stability of the different target protein structures in complexation with Valencene and respective reference drugs. The comparative stability assessment of three distinct neurodegenerative target proteins bound to Valencene and reference drugs further confirmed the strong modulatory potential of Valencene in comparison to the reference drugs.
Binding free energy changes (ΔGBFE) or thermodynamic stability
The binding free energy change (ΔGBFE) was used to assess the feasibility and spontaneity of the complex formation: the lower the negative value of ΔGBFE, the greater the stability of the protein–ligand system.25,26 The free energy changes of the adducts calculated for an equilibrated part of the trajectory (20 ns) are shown in Table 4. The best ΔGBFE was observed for the MAOB–Valencene complex with −25.18 ± 2.30 kcal mol–1. Similarly, negative binding free energy changes of −15.17 ± 3.44 and −18.98 ± 2.38 kcal mol–1 were observed for the AChE–Valencene and HTT–Valencene complexes respectively. ΔGBFE of the MAOB–Valencene and HTT–Valencene complexes were detected comparatively greater in value than that of MAOB–Selegiline and HTT–Ciproxifan complexes respectively. The result supported the better stability and spontaneity of the complex formation reaction of Valencene with MAOB and HTT proteins compared to those of the reference drugs (Table 4). However, the AChE–Rivastigmine complex was found to be formed with the release of slightly higher energy than in the formation of the AChE–Valencene complex. So, ΔGBFE for all three complexes suggested the spontaneous nature of complex formation and a more favourable forward complex formation reaction of Valencene with MAOB and HTT proteins compared to those of the complexes of the reference drugs.
| Complexes | ΔEVDWAALS | ΔEEL | ΔEPB | ΔENPOLAR | ΔGGAS | ΔGSOLV | ΔGBFE | |
|---|---|---|---|---|---|---|---|---|
| MAOB–Valencene | −37.81 ± 1.65 | −1.14 ± 0.45 | 16.78 ± 1.58 | −3.01 ± 0.05 | −38.95 ± 1.71 | 13.77 ± 1.58 | −25.18 ± 2.30 | |
| AChE–Valencene | −28.94 ± 1.88 | −0.36 ± 1.12 | 17.31 ± 3.47 | −3.17 ± 0.09 | −29.31 ± 2.23 | 14.14 ± 3.44 | −15.17 ± 3.44 | |
| HTT–Valencene | −29.87 ± 2.19 | −0.11 ± 0.86 | 13.87 ± 1.78 | −3.08 ± 0.09 | −29.76 ± 2.17 | 10.79 ± 1.76 | −18.98 ± 2.38 | |
| MAOB–Selegiline | −29.44 ± 1.63 | −2.34 ± 2.24 | 21.51 ± 3.03 | −3.64 ± 0.08 | −52.43 ± 6.68 | 31.01 ± 5.53 | −21.43 ± 6.67 | |
| AChE–Rivastigmine | −34.87 ± 2.11 | −17.80 ± 2.63 | 34.94 ± 3.71 | −3.89 ± 0.09 | −52.66 ± 3.11 | 31.30 ± 3.69 | −21.37 ± 3.21 | |
| HTT–Ciproxifan | −29.44 ± 1.63 | −2.34 ± 2.24 | 21.51 ± 3.03 | −3.75 ± 0.09 | −31.78 ± 2.97 | 18.45 ± 3.02 | −13.33 ± 2.98 |
Drug-likeness and toxicity
The pharmacokinetic and pharmacodynamic characteristics of Valencene were predicted by in silico tools to evaluate its drug-likeness and suitability for human consumption, as depicted in Table 5. From the prediction of absorption, metabolism, excretion, distribution and toxicity (ADMET), it was observed that the Valencene molecule belongs to toxicity class 5 and obeys the Lipinski’s rule of 5 (RO5), suggesting a probability for drug-likeness. The compound did not show any sign of immunotoxicity, carcinogenicity, mutagenicity, cytotoxicity or hepatotoxicity but exhibited skin sensitisation and neurotoxicity, analogous to the reference drugs, selegiline and rivastigmine. The blood–brain barrier (BBB) plays a very crucial role in the treatment of neurodegenerative disorders, as a majority of molecules encounter difficulties in crossing this barrier.27 The hit candidate and both reference drugs demonstrated the potential to cross the BBB, indicating their capability to function as brain-targeting drugs. The metabolic activity of Valencene was similar to that of the drugs and exhibited no hERG-blocking properties, implying that the compound does not induce fatal ventricular arrhythmia. The compound demonstrated high intestinal absorption, like the reference drugs, representing effective passage of the compound through the intestinal cell membranes suitable as an oral formulation.28 The total clearance of the compound was almost similar to the clearance of the reference drug, suggesting its potential to be used as a drug.
| ADMET parameters | Valencene and reference drugs | |||
|---|---|---|---|---|
| Valencene | Selegiline | Rivastigmine | ||
| Toxicity class | 5 | 4 | 4 | |
| Lipinski’s rule of 5 (RO5) | Yes | Yes | Yes | |
| Immunotoxicity | No | No | Yes | |
| Hepatotoxicity | No | No | No | |
| Carcinogenicity | No | No | No | |
| Mutagenicity | No | No | No | |
| Neurotoxicity | Yes | Yes | Yes | |
| Cytotoxicity | No | No | No | |
| Skin sensitisation | Yes | Yes | No | |
| hERG I inhibitor | No | No | No | |
| hERG II inhibitor | No | No | No | |
| CYP2D6 substrate | No | No | No | |
| CYP3A4 substrate | Yes | No | Yes | |
| CYP1A2 inhibitor | No | Yes | No | |
| CYP2C19 inhibitor | No | No | No | |
| CYP2C9 inhibitor | Yes | Yes | No | |
| CYP2D6 inhibitor | No | No | No | |
| CYP3A4 inhibitor | No | No | No | |
| BBB permeability | Yes | Yes | Yes | |
| Skin permeability | Low | Low | High | |
| Caco2 permeability | High | High | High | |
| Intestinal absorption | High | High | High | |
| Total clearance log (mL min–1 kg–1) | 1.205 | 1.007 | 0.557 | |
The comparative evaluation of toxicity and pharmacokinetics of the Valencene compound with reference drugs revealed its less toxic nature along with its potential to treat neurodegenerative disorders. The ADMET results suggested further exploration of the hit compound through experimental trials, in vivo and in vitro, to assess its suitability for human consumption.
Conclusion
Among the 42 compounds identified from the GC-MS analysis of the essential oil of Psidium guajava L., the in silico approach revealed Valencene as a potent neurodegenerative inhibitor in comparison to the reference drugs (Selegiline, Rivastigmine and Ciproxifan). The compounds exhibited strong thermodynamic and geometric stability with all the proteins as adducts and could possibly inhibit the normal functioning of the enzyme possibly leading to the treatment of neurodegenerative diseases. ADMET analysis suggested that Valencene could be a drug-like candidate with safe toxicity levels. However, further in vivo and in vitro experiments are necessary to validate the in silico findings. Hence, plant-derived compounds could be employed in addressing neurodegenerative disorders.
Experimental
Collection of plant samples and extraction of essential oil
The leaves of Psidium guajava L. were collected from Kathmandu, Nepal, and verified by the National Herbarium and Plant Laboratories, Lalitpur, Nepal. The essential oil from the leaves was extracted through hydrodistillation using a Clevenger-type apparatus.29
GC-MS analysis
Gas chromatography–mass spectrometry (GC-MS) experiments were performed using a GCMS-QP 2010 instrument. Helium was used as the carrier gas, passing through an RTX-5MS column of dimension 60 m × 0.32 mm × 0.25 μm. The temperature profile included a gradual increase from 80 to 300°C, with respective hold times for the start and end of 2.0 and 5.0 min. Throughout the analysis, the ion source and interface were consistently maintained at 200 and 250°C respectively, and the compound identification relied on MS comparison using the Flavor and Fragrance Natural and Synthetic Compounds (FFNSC, ver. 4.0) library.30
In silico approach
A database composed of 42 ligands, obtained from the GC-MS analysis of the oil of P. guajava was prepared. The 3-D structure of the ligand was downloaded from the PubChem database in SDF format31 (see https://pubchem.ncbi.nlm.nih.gov/) and the Avogadro software (ver. 1.2.0, see https://avogadro.cc/) was used to check their bond order and molecular structure.32 The 3-D crystalline structures of the proteins, Human monoamine oxidase B (PDB ID: 4A79, X-ray crystallographic resolution: 1.89 Å, expression system: Komagataella pastoris), Human acetylcholinesterase (PDB ID: 7E3H, X-ray crystallographic resolution: 2.45 Å, expression system: Homo sapiens) and Human histamine receptor H3R (PDB ID: 7F61, X-ray crystallographic resolution: 2.60 Å, expression system: Spodoptera frugiperda) were obtained in pdb format from the RCSB protein data bank33 (see https://www.rcsb.org/). The proteins were then cleaned using the PyMOL program (ver. 2.5.2, see https://www.pymol.org/34) and saved as an apo structure.
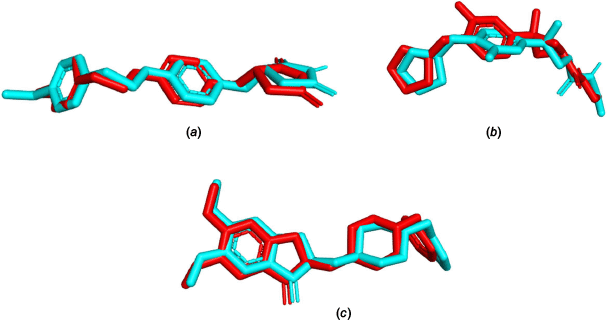
The molecular docking calculation was carried out using the DockThor server (see https://dockthor.lncc.br/) to determine the best binding pose of the ligand within the receptor protein.35 The docking program was selected because of its capability to perform numerous docking calculations quickly, being freely available and for its ability to reproduce the results. Different control parameters were used for the docking of ligands with different proteins. For the protein 4A79, box coordinates (51, 157, 31), box size (15, 15, 15), discretisation (0.16), population size (750), numbers of evaluations (1,000,000) and numbers of runs (24) were selected. The box size (16, 16, 16), box coordinates (−43, 36, −32), discretisation (0.17), population size (750), number of runs (24) and evaluations (1,000,000) were selected for protein 7E3H. Similarly, box coordinates (−19, 50, −1), box size (15, 15, 15), discretisation (0.16), population size (750), number of runs (24) and evaluations (1,000,000) were used for protein 7F61. The validation of the docking protocol was done by obtaining the RMSD of less than 2 Å for each (1.420, 0.669 and 1.666 Å for MAOB: 4A79, AChE: 7E3H, and HTT: 7F61 proteins respectively) through the superimposition of co-crystallised ligands in the crystalline complexes with re-docked ligands as shown in Fig. 7.
Molecular dynamics simulations of the best ligand–protein complexes were carried out using the GROMACS program (ver. 2021.2, see http://www.gromacs.org/)36 and the SwissParam server (see https://www.swissparam.ch/).37 An adduct in a triclinic box system of 10-Å spacing was solvated using the TIP3P water model.38 An isotonic solution of NaCl (0.15 M) was used and Na+ or Cl– ions were added to neutralise the system. It was equilibrated at the temperature of 310 K in four stages i.e. two NVT equilibria and two final NPT equilibria (500 ps each). Other parameters were adopted from recent literature39 and the final production run was conducted for 100 ns with a step size of 2 fs.
The binding free energy changes of the protein–ligand complex are given by the mathematical equation40:
where ΔGcomplex is the free energy of protein–ligand complex, ΔGprotein is free energy of protein, and ΔGligand is free energy of ligand.
The absorption, metabolism, excretion, distribution and toxicity of the compound was assessed through the servers ProTox (ver. 3.0, see https://tox.charite.de/protox3/),41 pkCSM (see https://biosig.lab.uq.edu.au/pkcsm/)42 and SwissADME (see http://www.swissadme.ch/).43
Supplementary material
The chromatogram, mass spectra, chemical structure of compounds indentified from GC-MS analysis (Supplementary Fig. S1–S3) and binding affinity table (Supplementary Table S1) are available as supplementary material. Supplementary material is available online.
Data availability
The data that support this study are available in the article and accompanying online Supplementary material.
Acknowledgements
The authors acknowledge the Department of Plant Resources, Kathmandu, Nepal, for the GC-MS experiments.
References
1 Amor S, Puentes F, Baker D, Van Der Valk P. Inflammation in neurodegenerative diseases. Immunology 2010; 129(2): 154-169.
| Crossref | Google Scholar | PubMed |
2 Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med 2004; 10(10): 1055-1063.
| Crossref | Google Scholar | PubMed |
3 Prince M, Wimo A, Guerchet M, Gemma-Claire A, Wu YT, Prina M. World Alzheimer Report 2015: the Global Impact of Dementia – an analysis of prevalence, incidence, cost and trends. Vol. 84. Alzheimer’s Disease International; 2015. Available at https://www.alzint.org/u/WorldAlzheimerReport2015.pdf
4 Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, et al. Alzheimer’s disease. Lancet 2021; 397(10284): 1577-1790.
| Crossref | Google Scholar | PubMed |
5 GBD 2016 Dementia Collaborators. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 2019; 18(1): 88-106.
| Crossref | Google Scholar | PubMed |
6 Lang AE, Lozano AM. Parkinson’s disease. N Engl J Med 1998; 339(16): 1130-1143.
| Crossref | Google Scholar | PubMed |
7 Smalley JL, Breda C, Mason RP, Kooner G, Luthi-Carter R, Gant TW, et al. Connectivity mapping uncovers small molecules that modulate neurodegeneration in Huntington’s disease models. J Mol Med 2016; 94(2): 235-245.
| Crossref | Google Scholar | PubMed |
8 Roos RA. Huntington’s disease: a clinical review. Orphanet J Rare Dis 2010; 5: 40.
| Crossref | Google Scholar | PubMed |
9 Talesa VN. Acetylcholinesterase in Alzheimer’s disease. Mech Ageing Dev 2001; 122(16): 1961-1969.
| Crossref | Google Scholar | PubMed |
10 Riederer P, Danielczyk W, Grünblatt E. Monoamine oxidase-b inhibition in Alzheimer’s disease. Neurotoxicology 2004; 25(1–2): 271-277.
| Crossref | Google Scholar | PubMed |
11 Zhou G, Miura Y, Shoji H, Yamada S, Matsuishi T. Platelet monoamine oxidase B and plasma β-phenylethylamine in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2001; 70: 229-231.
| Crossref | Google Scholar | PubMed |
12 Kohli H, Kumar P, Ambasta RK. In silico designing of putative peptides for targeting pathological protein Htt in Huntington’s disease. Heliyon 2021; 7(2): e06088.
| Crossref | Google Scholar | PubMed |
13 Singh SP. Guava (Psidium guajava L.). In: Yahia EM, editor. Postharvest Biology and Technology of Tropical and Subtropical Fruits. Woodhead Publishing Limited; 2011. pp. 213–246. doi:10.1533/9780857092885.213
14 Kumar M, Tomar M, Amarowicz R, Saurabh V, Nair MS, Maheshwari C, et al. Guava (Psidium guajava L.) leaves: nutritional composition, phytochemical profile, and health-promoting bioactivities. Foods 2021; 10: 752.
| Crossref | Google Scholar | PubMed |
15 Jeong CH, Jeong HR, Choi GN, Kwak JH, Kim JH, Park SJ, et al. Neuronal cell protective effects of hot water extracts from guava (Psidium guajava L.) fruit and leaf. Korean J Food Preserv 2011; 18(1): 124-129.
| Crossref | Google Scholar |
16 Aly SH, Eldahshan OA, Al-Rashood ST, Binjubair FA, El Hassab MA, Eldehna WM, et al. Chemical constituents, antioxidant, and enzyme inhibitory activities supported by in silico study of n-hexane extract and essential oil of guava leaves. Molecules 2022; 27(24): 8979.
| Crossref | Google Scholar | PubMed |
17 Neupane P, Dhital S, Parajuli N, Shrestha T, Bharati S, Maharjan B, et al. Exploration of anti-diabetic potential of Rubus ellipticus smith through molecular docking, molecular dynamics simulation, and MMPBSA calculation. J Nepal Phys Soc 2023; 9(2): 95-105.
| Crossref | Google Scholar |
18 Shrestha RLS, Panta R, Maharjan B, Shrestha T, Bharati S, Dhital S, et al. Molecular docking and ADMET prediction of compounds from Piper longum L. detected by GC-MS analysis in diabetes management. Mor J Chem 2024; 12(2): 776-798.
| Crossref | Google Scholar |
19 Shrestha RLS, Neupane P, Dhital S, Parajuli N, Maharjan B, Shrestha T, et al. Selected phytochemicals as potent acetylcholinesterase inhibitors: an in silico prediction. J Serbian Chem Soc 2024; 90: 187-200.
| Crossref | Google Scholar |
20 Shrestha RLS, Parajuli N, Neupane P, Dhital S, Maharjan B, Shrestha T, et al. A computational approach of anti-diabetic potential evaluation of flower and seed of Nyctanthes arbor tristis Linn. Turk Comp Theor Chem 2025; 9(1): 1-18.
| Crossref | Google Scholar |
21 Salo-Ahen OMH, Alanko I, Bhadane R, Alexandre AM, Honorato RV, Hossain S, et al. Molecular dynamics simulations in drug discovery and pharmaceutical development. Processes 2021; 9: 71.
| Crossref | Google Scholar |
22 Abdizadeh R, Hadizadeh F, Abdizadeh T. In silico analysis and identification of antiviral coumarin derivatives against 3-chymotrypsin-like main protease of the novel coronavirus SARS-CoV-2. Mol Divers 2022; 26(2): 1053-1076.
| Crossref | Google Scholar | PubMed |
23 Lobanov MY, Bogatyreva NS, Galzitskaya O V. Radius of gyration as an indicator of protein structure compactness. Mol Biol 2008; 42(4): 623-628.
| Crossref | Google Scholar |
24 da Fonseca AM, Caluaco BJ, Madureira JMC, Cabongo SQ, Gaieta EM, Djata F, et al. Screening of potential inhibitors targeting the main protease structure of SARS-CoV-2 via molecular docking, and approach with molecular dynamics, RMSD, RMSF, H-Bond, SASA and MMGBSA. Mol Biotechnol 2024; 66: 1919-1933.
| Crossref | Google Scholar | PubMed |
25 Cournia Z, Allen B, Sherman W. Relative binding free energy calculations in drug discovery: recent advances and practical considerations. J Chem Inf Model 2017; 57: 2911-2937.
| Crossref | Google Scholar | PubMed |
26 Dhital S, Parajuli N, Poudel M, Shrestha T, Bharati S, Maharjan B, et al. Spatial and energetic stability assessment of the adducts of phytocompounds of Piper longum L. with α-amylase by computational approach. Biointerface Res Appl Chem 2024; 14(7): 126.
| Crossref | Google Scholar |
27 Pardridge WM. Treatment of Parkinson’s disease with biologics that penetrate the blood–brain barrier via receptor-mediated transport. Front Aging Neurosci 2023; 15: 1276376.
| Crossref | Google Scholar | PubMed |
28 Shen J, Cheng F, Xu Y, Li W, Tang Y. Estimation of ADME properties with substructure pattern recognition. J Chem Inf Model 2010; 50(6): 1034-1041.
| Crossref | Google Scholar | PubMed |
29 Elyemni M, Louaste B, Nechad I, Elkamli T, Bouia A, Taleb M, et al. Extraction of essential oils of Rosmarinus officinalis L. by two different methods: hydrodistillation and microwave assisted hydrodistillation. ScientificWorldJournal 2019; 2019: 3659432.
| Crossref | Google Scholar | PubMed |
30 Pradhan S, Paudel HR, Maharjan R, Sharma K. Essential oils from six aromatic plants of Langtang National Park: insights on their chemical constituents via GC-MS analysis. Separations 2023; 10(1): 52.
| Crossref | Google Scholar |
31 Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem 2023 update. Nucleic Acids Res 2023; 51(D1): D1373-D1380.
| Crossref | Google Scholar | PubMed |
32 Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform 2012; 4(8): 17.
| Crossref | Google Scholar | PubMed |
33 Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res 2000; 28(1): 235-242.
| Crossref | Google Scholar | PubMed |
34 Yuan S, Chan HCS, Hu Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip Rev Comput Mol Sci 2017; 7: e1298.
| Crossref | Google Scholar |
35 Santos KB, Guedes IA, Karl ALM, Dardenne LE. Highly flexible ligand docking: benchmarking of the DockThor program on the LEADS-PEP protein–peptide data set. J Chem Inf Model 2020; 60(2): 667-683.
| Crossref | Google Scholar | PubMed |
36 Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, et al. Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015; 1–2: 19-25.
| Crossref | Google Scholar |
37 Zoete V, Cuendet MA, Grosdidier A, Michielin O. SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem 2011; 32(11): 2359-2368.
| Crossref | Google Scholar | PubMed |
38 Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys 1983; 79(2): 926-935.
| Crossref | Google Scholar |
39 Neupane P, Adhikari Subin J, Adhikari R. Assessment of iridoids and their similar structures as antineoplastic drugs by in silico approach. J Biomol Struct Dyn 2024; 1-16.
| Crossref | Google Scholar | PubMed |
40 Valdés-Tresanco MS, Valdés-Tresanco ME, Valiente PA, Moreno E. Gmx_MMPBSA: a new tool to perform end-state free energy calculations with GROMACS. J Chem Theory Comput 2021; 17(10): 6281-6291.
| Crossref | Google Scholar | PubMed |
41 Banerjee P, Kemmler E, Dunkel M, Preissner R. ProTox 3.0: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res 2024; 52(W1): W513-W520.
| Crossref | Google Scholar | PubMed |
42 Pires DEV, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem 2015; 58(9): 4066-4072.
| Crossref | Google Scholar | PubMed |
43 Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports 2017; 7(1): 42717.
| Crossref | Google Scholar | PubMed |