

Electrophilic fragment screening using native mass spectrometry to identify covalent probes for surface cysteines
Jack W. Klose A B # , Yezhou Yu A B # , Giovanna Di Trapani B , Kathryn F. Tonissen A B , Louise M. Sternicki A B * and Sally-Ann Poulsen A B *
A B # , Yezhou Yu A B # , Giovanna Di Trapani B , Kathryn F. Tonissen A B , Louise M. Sternicki A B * and Sally-Ann Poulsen A B *
A
B
# Co-first authors
Handling Editor: Mibel Aguilar
Abstract
Covalent chemical probes form a covalent bond with a target protein of interest to elicit an effect and methods to identify and characterise them are needed. We developed a native mass spectrometry (nMS) method to screen an electrophilic covalent fragment library and identified specific novel binders for the surface exposed cysteine residues of carbonic anhydrase III (CA III). The nMS method was extended to determine the site of protein modification and measure simultaneous binding of an active site noncovalent inhibitor and covalent fragment hit, which is not possible with intact denaturing MS. This study demonstrates the utility of using nMS and the advantages when compared to intact denaturing MS for the discovery and characterisation of new covalent ligands.
Keywords: acrylamide, carbonic anhydrase, carbonic anhydrase inhibitor, chloroacetamide, covalent drug discovery, cysteine, electrophilic fragments, fragment based drug discovery, native mass spectrometry, thiocyanate.
Introduction
Covalent small molecule drugs form a covalent bond with a target protein of interest to elicit the desired therapeutic response. Their chemical structure comprises a mildly reactive electrophilic warhead that forms a covalent bond with specific nucleophilic amino acid residues of the protein target.1 Although milestones in covalent drug discovery and development span more than one century, the modern trend in medicinal chemistry is toward rationally designed covalent drugs in place of serendipitous discovery.1,2 This more deliberate approach has been accompanied by a striking increase in the number of FDA approved covalent drugs since 2010.1 This trend is underpinned by the multiple benefits provided by covalent drugs, including high potency and selectivity, low off-target effects and lower dosage. Covalent drugs also provide opportunities to target proteins considered undruggable, particularly those with rare or non-conserved nucleophilic amino acid residues.1,3 This is embodied by approval of Sotorasib in 2021 and Adagrasib in 2022, covalent drugs respectively comprising an acrylamide and 2-fluoroacrylamide warhead that selectively target the reactive Cys12 of KRAS(G12C). This cancer-specific mutant isoform had remained undruggable despite decades of conventional drug discovery efforts.4–8 Additional recently approved covalent drugs that selectively target cysteine residues include Nirmatrelvir and Ritlecitinib. Nirmatrelvir comprises a nitrile warhead and is an inhibitor of the main protease from SARS-CoV2 (by Cys145) that was approved by the FDA for emergency use treating COVID-19 in 2021.1,9 Ritlecitinib, comprises an acrylamide warhead that selectively targets JAK3 (by Cys909) and the equivalent cysteine residue in TEC kinases and was first approved by the FDA in 2023 for the treatment of severe alopecia areata.10,11
Contemporary approaches in covalent drug discovery proceed through either a ligand-first approach, where incorporating covalency into a known reversible ligand is pursued, or an electrophile-first approach, where screening commercial or proprietary electrophilic compound libraries (comprising fragments or lead-like molecules) is employed to identify a novel covalent ligand ‘hit’, often without structural bias.1 Indeed, the electrophile-first approach was employed in the development of Sotorasib, starting with covalent fragments.4–6 Fragments are molecules, typically <300 Da in molecular weight, that are not only good starting points for the hit-to-lead stage of drug development but also as chemical probes or tool compounds that are used to study the biology of a protein of interest, particularly where the protein is unligandable or ligands are unavailable, limited or suboptimal.12 The growing interest in covalent inhibitors and covalent chemical probes requires robust technologies for the identification and characterisation of covalent fragment hits. Covalent binders impart different practical considerations for screening when compared to noncovalent binders and there is a need for methods to characterise direct target engagement. Herein, we demonstrate the application of native mass spectrometry (nMS) for covalent fragment screening. Our method provides a workflow that can assess protein binding, protein specificity and identification of the site of protein modification (utilising mutant proteins). We also demonstrate that nMS can measure the simultaneous binding of a protein to a classical active site noncovalent ligand and a covalent fragment – an outcome not possible with denaturing MS methods.
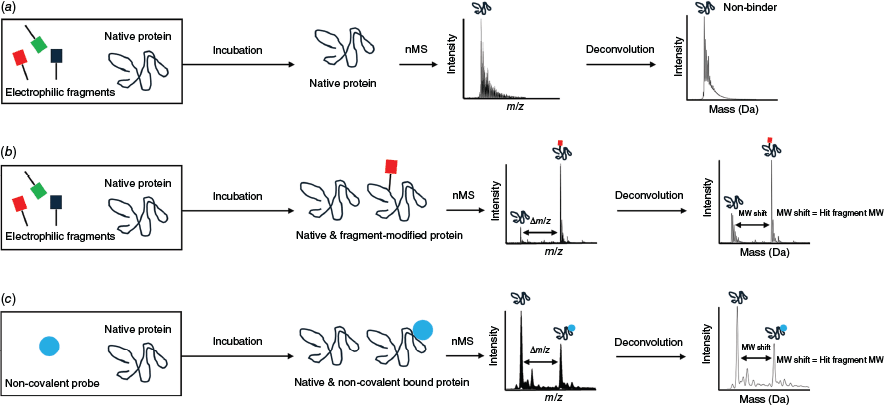
Intact denatured mass analysis has been used for electrophilic fragment screening across different human and pathogen protein target classes, including proteases and kinases.1,13–15 Following incubation of the protein of interest with a fragment (either a single fragment or fragment pools), the secondary, tertiary and any quaternary structures of a protein are disrupted prior to acquisition of a mass spectrum of the denatured protein. Covalent fragment binding is identified by the mass information in modified versus unmodified protein peaks in the deconvoluted mass spectrum.16 By contrast, nMS maintains the protein structure by using sample and MS instrument parameters that preserve the protein’s native-like folded structure and is established for noncovalent fragment screening (reviewed in Sternicki and Poulsen17,18 and Poulsen19). Covalent fragment binding may be determined directly from the simpler nMS raw data (as there are much fewer charge states than for denatured proteins) by the difference in the higher m/z value for the modified protein compared to the lower m/z value for the unmodified protein,20,21 or deconvoluted in the same way as intact denatured MS (Fig. 1). Covalent fragment binding is unambiguous with both approaches as the covalent bond remains intact and quantitative binding analysis is possible. The signal intensities for the unmodified and covalent ligand-modified protein peaks are likely to be an accurate reflection of the solution-based concentrations, due to similar MS response factors.22–26 Although the methods can identify multiple covalent modifications (e.g. multiple binding of the same fragment at different sites or binding of different fragments from a pool), only nMS provides the opportunity to identify noncovalent ligand binding. This is powerful as it enables simultaneous binding of orthosteric inhibitors and covalent ligands to be measured, providing important details to better guide development. It may also identify fragments where there is structural complementarity to the target protein but where an adjustment to the covalent warhead is needed to optimise covalent bond formation. We therefore sought to establish nMS as a robust new method to screen electrophilic fragment libraries.
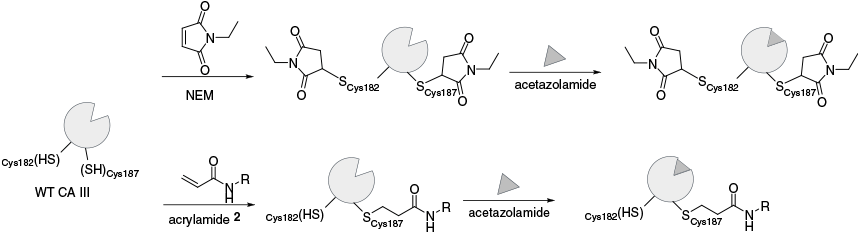
Native mass spectrometry (nMS) workflow for electrophilic fragment screening of pooled fragments against a protein of interest. (a) With no covalent fragment binding, the mass spectrum is unchanged. With covalent binding (b) or noncovalent binding (c) of a fragment, an additional peak corresponding to the mass of the protein plus that fragment is observed. For (b) and (c), the mass difference between the protein only and protein-fragment peaks (labelled Δm/z for raw mass spectrum or molecular weight, MW, shift for deconvoluted mass spectrum) allows identification of the fragment, with fragment mass as proxy for fragment identity calculated using MW = Δm/z × z.
Results and discussion
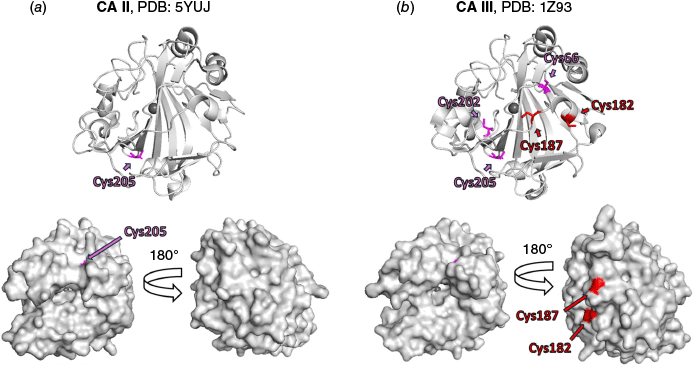
Carbonic anhydrases (CA, EC 4.2.1.1) catalyse the reversible hydration of carbon dioxide to give bicarbonate and a proton, playing an important pH regulatory role in health and disease.27 In humans, there are 12 catalytically active isozymes, with CA III the least active (kcat 1.3 × 104 s−1) and the least studied.28 CA III is, however, implicated to play a role in various diseases, including cancer and has an intriguing antioxidant function in cells by providing protection against cellular oxidative stress conditions.29,30 The three-dimensional protein fold of CA III is highly similar to the fold of CA II, the most active CA isozyme (kcat 1.4 × 106 s−1),28 with CA II and CA III sharing 62% sequence homology (see Fig. 2, 3 and S1).31 Although cysteine is one of the least abundant amino acids in proteins (1.9% abundance),32 its presence can signify an important functional role.32,33 CA III has five cysteine residues (C66, C182, C187, C202 and C205, Fig. 3 and S1), with structural studies showing that two are localised on the protein surface (C182 and C187, Fig. 2) and experience redox-regulated modifications, including glutathionylation,34 which helps protect cells from oxidative stress.29 There are currently no ligands that target these two cysteines which enable researchers to better understand the function of CA III or its potential as a therapeutic target,34 with site-directed mutagenesis the only available method to study them. The reported human CA III crystal structures (PDB IDs: 1Z93, 1Z97, 2HFW, 3UYN and 3UYQ) have C182 and C187 mutated to serine residues to assist crystallisation (our attempts to crystallise CA III without these mutations were not successful, data not shown). To the best of our knowledge, CA proteins have not been intentionally screened against electrophilic fragments that target cysteine using nMS; however, covalent binding to CA I and CA II has been reported when screening small molecule libraries.35,36 We thus sought to develop an electrophilic fragment screening method using nMS to identify selective binders for C182 or C187 in CA III. CA II has one cysteine (C205), a buried residue also found in CA III, providing a protein control for counter-screening to support the discovery of specific and selective covalent probes for the surface cysteine residues of CA III. Although not in the scope of this study, the CA isoform sequence alignment shows that mitochondrial CA VB has cysteine residues that align with C182 and C187 of CA III, mitochondrial CA VA has a cysteine residue that aligns with C187, and CA VII has a cysteine residue that aligns with C182. CA VII is reported to have a protective role against oxidative damage37 but there is no reported information on the role of these cysteines in the two mitochondrial CAs (CA VA and VB).
Comparison of protein X-ray crystal structures of (a) CA II (PDB 5YUJ), and (b) double mutant CA III (PDB 1Z93, C182S and C187S). CA III contains three buried cysteines (C66, C202 and C205) and CA II contains one buried cysteine (C205), highlighted in pink; the relative per residue solvent accessibility surface area is 0%, calculated using PyMoL (ver. 1.8, Schrodinger LLC, see https://www.pymol.org/). CA III contains two predicted surface exposed cysteines (C182 and C187) highlighted in red; the relative per residue solvent accessibility is respectively 29 and 25%, calculated using PyMoL. For ease of representation, the double mutant Ser residues of (b) are labelled Cys. The figures were prepared using PyMol.
Alignment of the amino acid sequences of catalytically active human carbonic anhydrase (CA) enzymes, focusing on the surface cysteine-containing sequence region. The two surface-exposed cysteine residues in CA III (C182 and C187) and the corresponding aligned cysteines in other CA isoforms are highlighted in red. The buried cysteine residue (C205) in both CA II and CA III is highlighted in blue. Other cysteine residues are highlighted in yellow. The human CA sequences used are CA I: NP_001122303.1, CA II: NP_000058.1, CA III: NP_005172.1, CA IV: NP_000708.1, CA VA: NP_001730.1, CA VB: NP_009151.1, CA VI: NP_001206.2, CA VII: NP_005173.1, CA IX: NP_001207.2, CA XII: NP_001209.1, CA XIII: NP_940986.1 and CA XIV: NP_036245.1, with alignment performed using the MUSCLE (MUltiple Sequence Comparison by Log-Expectation) algorithm in Snapgene.

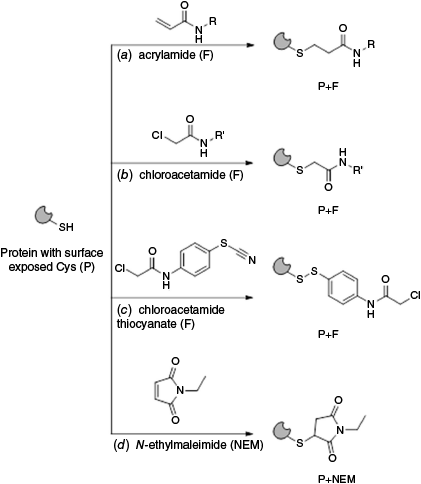
The Electrophilic Covalent Probes library (960 compounds), supplied by Enamine and co-developed by the Weizmann Institute of Science and XChem group at Diamond Light Source,13 is curated toward the discovery of specific ligands for cysteine-containing proteins. The library comprises cysteine-reactive acrylamide (~24%) and chloroacetamide (~76%) warheads on the fragments, with the expected covalent modification reactions shown in Fig. 4a, b respectively. Thiol-reactivity against 10 cysteine-containing proteins was evaluated during library development13 and the library has since been screened with other targets.38 We employed nMS to screen this electrophilic fragment library (4 equiv) against wildtype (WT) CA III. N-Ethylmaleimide (NEM), a promiscuous alkylating reagent for protein thiol moieties at pH 6.5–7.5 (Fig. 4d) was employed as a positive control (4 equiv), whereas DMSO was employed as a negative control. Covalent ligand binding was identified as previously described by us.20 NEM was observed to covalently modify two sites of WT CA III (presumably the surface exposed cysteines C182 and C187) with rapid binding of the first ligand and binding of the second ligand increasing in a time dependent manner (18.1% at 1 h, 28.7% at 4 h and 43.5% at 16 h, data not shown). Where putative hit fragments were identified or where data were ambiguous, the fragments were subjected to a secondary screen. Twelve fragments 1–12 were confirmed as covalent binders of WT CA III (1.25% hit rate) (Fig. 5a) each with one ligand bound to the protein. With respect to quality control of the electrophilic fragment library, there were ~1.7% of samples assessed as ‘bad wells’ in the primary nMS screen, where binding to WT CA III was observed but the mass of the modified protein was inconsistent with the fragments’ chemical structure, indicative of potential degradation of fragments or impurities.
Covalent modification of protein surface thiols with cysteine directed electrophilic fragments (F) comprising (a) acrylamide and (b) chloroacetamide covalent warheads. (c) A putative covalent modification of protein thiol with dual thiocyanate–chloroacetamide fragment (F). (d) Covalent protein modification with promiscuous cysteine alkylating reagent, N-ethyl maleimide (NEM).
(a) Structure of confirmed electrophilic fragments 1–12 that bound covalently to WT CA III using nMS. (b) Structure of non-binding hit analogues 13–20 identified by cross referencing hits against the complete fragment library.
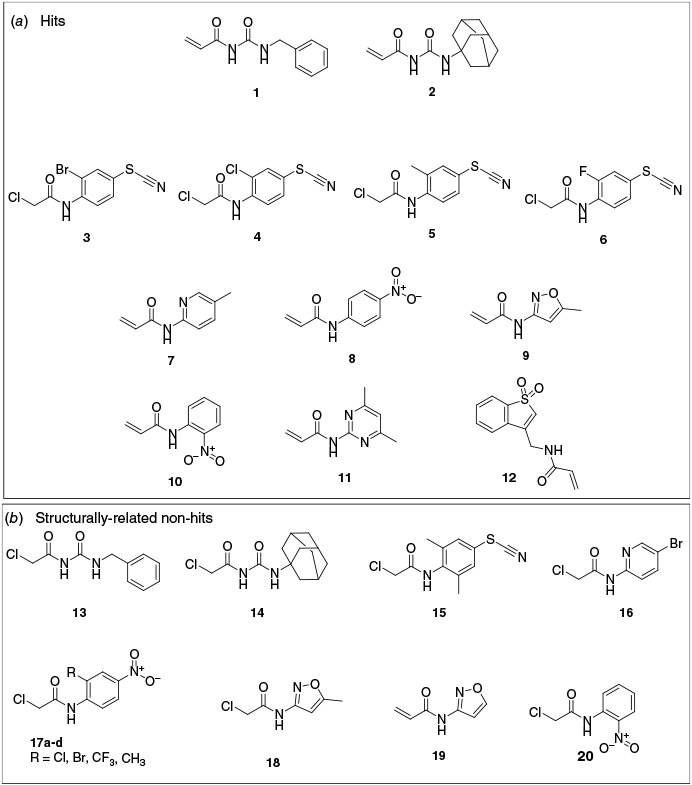
Hit fragments for WT CA III comprised three pharmacophores: (i) acrylamides with a urea functionality and a bulky hydrophobic group (1 benzyl, 54.5% bound and 2 adamantyl, 44.0% bound); (ii) chloroacetamides with substituted phenyl thiocyanates (3 bromo, 29.9% bound; 4 chloro, 31.9% bound; 5 methyl, 25.9% bound and 6 fluoro, 19.9% bound); and (iii) heteroaromatic acrylamides 7–12 (11.5–26.8% bound) (Fig. 5a, S2, and Tables 1, S1 and S2). All hits bound as a single adduct, modifying only one cysteine residue (i.e. no double labelling as for NEM). The 12 hits are inclusive of all fragments that labelled CA III to any measurable extent (i.e. there was no minimum percentage labelling threshold applied) and that also met the following criteria: (1) hit fragment from nMS screening in pools (of four fragments) was validated and confirmed as a hit when re-screening as a single compound; (2) the measured mass shift for the hit fragment was consistent with the mass shift expected for covalent binding to the warheads or isothiocyanate moiety. Hits 1, 2 and 7–12 bound with the expected mass for covalent addition of the acrylamide fragment (i.e. mass shift equal to the fragment molecular weight, MW), with covalent binding confirmed by adjustment of MS parameters that are expected to disrupt noncovalent binders (FT-ICR MS skimmer 1 voltage increased from 30 to 120 V, UHMR MS desolvation voltage increased from −15 to −50 V) (Supplementary Fig. S3). Fragments 3–6 did not bind with the expected mass loss for a chloroacetamide (−36.5 Da) but instead with a 26-Da loss that was attributed to a putative reaction of the thiocyanate moiety of 3–6 with a cysteine thiol to form a disulfide (Fig. 4c). Only a single adduct with a mass shift of −26 Da was observed for fragments 3−6, suggesting a high specificity (>99%) for electrophilic reactivity from the isothiocyanate moiety over the chloroacetamide warhead. Although further investigation would be required to confirm this hypothesis, there is literature precedent for the thiocyanate–thiol reaction that contrasts with the covalent-reversible reaction of the isothiocyanate warhead (which reacts by the carbon centre with protein thiol groups).39,40 The two‐step mechanism of covalent binding, whereby the fragment initially binds to the target reversibly (mediated by noncovalent and specific interactions between the fragment and protein binding site) and then the binding conformation of the fragment directs the electrophilic warhead (in this case the thiocyanate) to react covalently with a proximal cysteine, suggests this alternative reaction mechanism is feasable.41,42
| Compound | Primary screen | Primary screen | Hit validation | Counter-screen | |
|---|---|---|---|---|---|
| Rep 1 | Rep 2 | ||||
| 1 | 82.6 | 74.1 | 54.5 | 0 | |
| 2 | 51.4 | 59.7 | 44.0 | 0 | |
| 3 | 17.4 | 40.2 | 29.9 | 0 | |
| 4 | <10 | 34.4 | 31.9 | 0 | |
| 5 | <10 | 34.4 | 25.9 | 0 | |
| 6 | <10 | 31.0 | 19.9 | 0 | |
| 7 | <10 | 24.5 | 26.8 | 0 | |
| 8 | 0 | 20.5 | 17.2 | 0 | |
| 9 | 0 | 20.1 | 19.4 | 0 | |
| 10 | 0 | 18.7 | 18.6 | 0 | |
| 11 | 0 | 16.8 | 11.5 | 0 | |
| 12 | 0 | 14 | 14.2 | 0 | |
| NEM | 77.7, 22.3A | 23.7, 76.3A | 64.1, 35.9A | 0 |
For Primary Screen Rep 1, Hit validation and Counter-screen, the nMS sample of CA protein was prepared in 10 mM of NH4OAc. For Primary Screen Rep 2, the nMS sample of CA III was prepared in 100 mM of NH4OAc. For Primary Screen Rep 1, FT-ICR MS was used for nMS screening. For Primary Screen Rep 2, Hit validation and Counter-screen, UHMR MS was used for nMS screening.
To assess hit selectivity for the CA III isoform, compounds 1–12 and NEM (control) were counter-screened against CA II (one buried cysteine, no surface cysteines). No binding to CA II was observed, indicating the hits were specific for surface exposed cysteines of CA III (Table 1). We also cross referenced hits 1–12 against the structures of the full fragment library and although all fragments in the library have an innate capacity to covalently modify cysteine, there was selectivity of the hit ligands 1–12 compared to the structurally related analogues 13–20 (Fig. 5b). This further supports a mechanism where noncovalent molecular interactions are a precursor for directing the fragment electrophile for specific covalent modification.42 Lastly to assess for off target protein promiscuous binding, we cross referenced hits 1–12 with the reported results for electrophilic fragment screening against the 10 cysteine containing proteins that guided the library’s development (Supplementary Table S3).13 We found that acrylamides 1 and 2 covalently modify the cysteines of at least three proteins including KRAS(G12C) (79 and 26% bound respectively), NUDT7 (35 and 79% bound respectively), NV3CP (hit 1 30% bound) and PBP(R504C) (hit 2 19% bound). The dual thiocyanate chloroacetamides 3–6 covalently modified one different protein each (hit 3 KRAS(G12C) 9% bound; hit 4 NNMT 38% bound; hit 5 PBP(R504C) 11% bound; hit 6 OTUB2 9% bound); however, data to assess if binding is by the thiocyanate moiety as we observed are not available. Heterocyclic acrylamide hits 7–12 covalently modified between two and four of the panel of 10 proteins tested (Supplementary Table S3). The promiscuity of the reactivity of hits 1–12 for these off target proteins was surprising given the library was described to have removed compounds found to be promiscuous binders (defined as those that label two or more proteins by >50% or three or more proteins by >30%) or highly reactive, in the screening of those 10 proteins.43 An alternative hypothesis is that the cysteines of those frequently hit proteins are too reactive to be specifically targeted with electrophilic fragments.
To identify the reactive cysteine of WT CA III we prepared three mutant CA III proteins (C182S, C187S, CDS, with both C182 and C187 as serine) that encompass the possible combinations of surface exposed cysteine residues mutated to serine. We then assessed NEM and fragment hits 1–12 (4 equiv, 16 h) for covalent binding using nMS (Tables 2, T1 and T2, Supplementary Fig. S4 and S5). NEM modified WT CA III (up to two NEM bound), C182S and C187S (with one NEM ligand bound) but did not modify CDS. This NEM binding data is consistent with the expected covalent modification of both surface exposed cysteines (C182 and C187) of WT CA III and no modification of the three buried cysteines (C66, C202 and C205). The fragment hits 1 and 2 covalently modified only WT CA III and C182S, providing evidence that C187 is selectively modified by these urea functionalised acrylamide fragments (with C182 unmodified) (Table 2). Similarly, heteroaromatic acrylamide hits 7–12 covalently modified only WT CA III and C182S, further evidence of selective modification of C187 only (Table 2). Although the dual thiocyanate–chloroacetamide fragments 3–6 did not modify CDS, they behaved differently towards the other mutants; specifically, fragments 3 and 5 covalently modified WT CA III, C182S and C187S (to a lesser extent), indicating these fragments could modify both surface cysteines through the thiocyanate moiety. By contrast, the dual thiocyanate chloroacetamide hits 4 and 6 covalently modified only WT CA III and C182S, indicative of a preference for modification of C187. In summary, all hits covalently and preferentially modified C187 over C182, with only two hits (3 and 5) modifying both C187 and C182, although C182 modification was to a much lesser extent.
| Compound | Percentage CA III bound | ||||
|---|---|---|---|---|---|
| WT | C182S | C187S | CDS | ||
| 1 | 54.5 | 52.1 | 0 | 0 | |
| 2 | 44.0 | 43.2 | 0 | 0 | |
| 3 | 29.9 | 25.0 | 9.7 | 0 | |
| 4 | 31.9 | 24.8 | 0 | 0 | |
| 5 | 25.9 | 20.2 | 7.1 | n.t. | |
| 6 | 19.9 | 14.9 | 0 | 0 | |
| 7 | 26.8 | 17.1 | 0 | n.t. | |
| 8 | 17.2 | 14.7 | 0 | n.t. | |
| 9 | 19.4 | 12.8 | 0 | n.t. | |
| 10 | 18.6 | 11.1 | 0 | n.t. | |
| 11 | 11.5 | 9.3 | 0 | n.t. | |
| 12 | 14.2 | 13.4 | 0 | n.t. | |
| NEM | 64.1 | 100 | 64.1 | 0 | |
| 35.9A | |||||
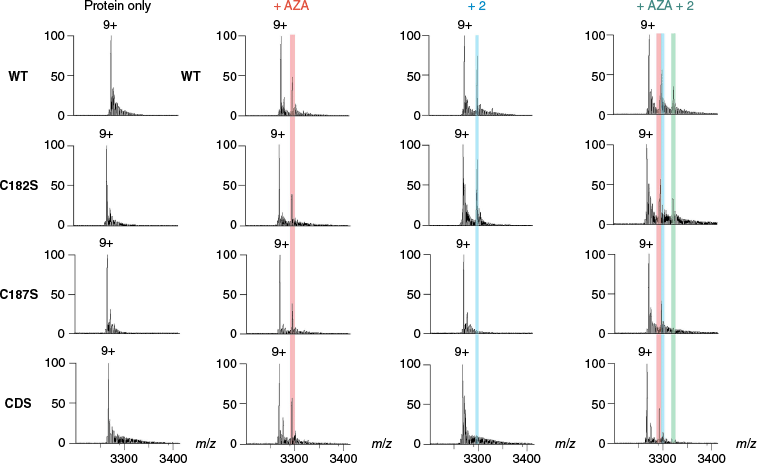
To assess the ability of nMS to bind with acetazolamide (2 equiv), a high affinity noncovalent binder of the CA II and CA III active sites,20 samples were incubated for 30 min and nMS data acquired (Table 3). The two proteins bound to acetazolamide at a similar level (34.5–37.5% binding), demonstrating that the active site architecture was similar even though the surface cysteine profile was different. Next, to assess the ability of nMS to identify simultaneous noncovalent orthosteric binding with acetazolamide and covalent binding with the electrophilic fragments, the proteins were incubated with NEM (4 equiv) as a positive control or a representative covalent hit (fragment 2, 4 equiv, 16 h), followed by incubation with acetazolamide (2 equiv, 30 min). Simultaneous binding of NEM and acetazolamide was observed for WT CA III (2× NEM bound) and mutants C182S (1× NEM bound) and C187S (1× NEM bound), but not mutant CDS (Tables 3, S4 and S5, and Supplementary Fig. S6 and S7). Simultaneous binding of 2 and acetazolamide was observed for WT CA III (1× 2 bound) and mutant C182S (1× 2 bound), but not for mutant C187S or CDS (Tables 3, S4 and S5, and Fig. 6, S8). Collectively, this provides evidence that both C187 and C182 may be modified by nonspecific alkylation with NEM but that there is specific covalent modification of C187 over C182 with the electrophilic fragment 2. It also confirms that the covalent protein modifications do not affect orthosteric binding of acetazolamide. Additionally, as expected, CA II did not bind NEM or the electrophilic fragment 2 and bound only to acetazolamide (Table 3).
 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Percentage P binding | Percentage P binding | Percentage P binding | ||||||||||
| Control samples | With combined NEM and AZA | With combined 2 and AZA | ||||||||||
| Protein (P) | AZA | NEM | 2 | Unbound | AZA | NEM | NEM + AZA | Unbound | AZA | 2 | 2 + AZA | |
| WT CA III | 34.5 | 94.9 | 40.7 | 0 | 0 | 43.6 | 23.9 | 42.3 | 16.5 | 25.1 | 16.1 | |
| CA III C182S | 35.1 | 100 | 45.4 | 0 | 10.9 | 60.4 | 28.7 | 41.8 | 13.9 | 28.3 | 16.0 | |
| CA III C187S | 35.5 | 60.8 | 0 | 25.5 | 21.0 | 33.3 | 19.8 | 63.1 | 36.9 | 0 | 0 | |
| CA III CDS | 37.5 | 0 | 0 | 66.2 | 33.8 | 0 | 0 | 64.1 | 35.9 | 0 | 0 | |
| WT CA II | 36.0 | 0 | 0 | 45.2 | 54.8 | 0 | 0 | 47.9 | 52.1 | 0 | 0 | |
Scheme is representative of the analyses. NEM and NEM+AZA binding percentage includes the sum of the single and double labelled binding for WT CA III (i.e. both one and two cysteines covalently modified).
Conclusion
Electrophilic fragments are an important class of fragments that form a covalent bond with protein targets, providing powerful tools that may be developed as chemical probes to explore the biology of targets. This is particularly relevant for target validation (or off-target identification) and as starting points for drug discovery programmes. In this study, we developed a new screening method using nMS to efficiently identify electrophilic fragment hits, identifying 12 selective electrophilic fragments for CA III across three pharmacophores. This is important as there is a lack of high-quality and specific chemical tools for CA III, providing a barrier to establishing the biological role(s) of this enzyme. Using nMS with mutant CA III proteins, we determined that C187 is the preferred site of fragment modification of the two possible surface cysteines (C182, C187). This finding will allow the attention of future efforts to understand the biology of CA III to focus on targeting C187 for hit-to-lead development. Additionally, the conserved homology of C187 with mitochondrial CAs VA and VB may be investigated to identify cross-reactivity of the identified hit probes for these CAs. nMS provided the opportunity to assess simultaneous orthosteric (noncovalent) binding and covalent binding to CA III to assess the impact of covalent surface cysteine modification on active site binding, which is not possible with denaturing MS methods. This general capability of nMS could greatly expand the capacity to develop functional covalent chemical probes where quantitative concurrent binding information is important.
nMS samples are analysed directly from solution; there is no requirement for protein crystallisation or protein modification as is needed for alternative biophysical methods (e.g. X-ray crystallography, surface plasmon resonance). This is significant as there is a vast number of proteins that are too flexible or too dynamic to crystallise or that are not amenable to modification, as it interferes with function or structure. Furthermore, the nMS method could enable screening of electrophilic fragments at scale, with the stoichiometry and identity of fragments determined from mass data. As nMS is a target agnostic analytical method, we anticipate it will find broad applications in electrophilic chemical probe discovery and drug discovery for protein targets. Continued development of the nMS method may aid identification of novel functional cysteines and guide the discovery of chemical probes for those proteins. There is potential to elaborate the nMS method to alternative nucleophilic amino acids beyond cysteine, or alternative electrophilic warheads, enabling a comprehensive range of covalent chemistries available to modify proteins.
Experimental
Recombinant protein expression and purification
Recombinant human wildtype (WT) CA II and WT CA III were expressed and purified as previously described.20,44,45 Three mutated variants of CA III, C182S, C187S and CDS, were produced, whereby cysteine residues at positions 182 and 187 were individually mutated to serine to give C182S CA III and C187S CA III respectively, whereas CA III CDS carried both mutations, replacing both cysteines (C182 and C187) with serine. These point mutations were introduced by IVA cloning46 and the proteins expressed and purified using the same method described for WT CA III.20
Native mass spectrometry protein sample preparation
CA proteins (WT CA III, CA III mutants (C182S, C187S and CDS) and WT CA II) were concentrated using a Vivaspin 20 ultrafiltration unit (10 kDa MWCO, Sartorius) and then buffer exchanged into 10 mM of NH4OAc, pH 7.0 (unless otherwise stated) using a Nap-5 column (Cytiva). CA protein concentration was determined by measuring the absorption at 280 nm and considering the proteins’ calculated extinction coefficients (WT CA III, C182S, C187S and CDS 55,920 M−1 cm−1; WT CA II 50,420 M−1 cm−1). The protein concentration was adjusted to 5 µM with 10 mM of NH4OAc, pH 7.0 and the protein samples stored at −80°C until used. CA III proteins (WT, C182S, C187S and CDS) were supplemented with equimolar ratios of Zn2+ (by way of zinc acetate) prior to addition to fragments for nMS analysis. WT CA II was saturated with bound Zn2+ following expression and purification.
Electrophilic covalent probes compound library screening and binding studies by nMS
For primary screening the Enamine Electrophilic Covalent Probe Library (957 compounds, 3 compounds were not soluble) was provided by Compounds Australia, Griffith University.47 The compounds were stored at 10 mM in 100% DMSO and plated in pools of 4 compounds per well (40 nL each 10-mM compound) in nMS-compatible 96-well plates (BioRad Hard-Shell 96-well PCR plates #HSP9601). Column 1 was plated with 160 nL of DMSO as a negative control and column 2 was plated with NEM (40 nL, 10 mM, backfilled with DMSO to a total volume of 160 nL) as a positive control for cysteine covalent modification. This provided positive and negative controls for every 10 wells (40 compounds) to monitor consistency of the nMS screening. WT CA III protein (5 µM, 20 µL) was added to each assay plate well to yield samples with 1 equiv protein and 4 equiv of each fragment (0.79% DMSO). Samples were incubated in the dark at room temperature for ~16 h prior to nMS analysis.
Selected hit fragments and compounds with ambiguous results from the primary screen were plated individually (40 nL, 10 mM, backfilled with DMSO to 160-nL total volume) in nMS-compatible 96-well plates for hit validation, counter-screening against WT CA II and screening against mutant CA III proteins. Columns 1 and 2 were prepared similarly to the primary screen with negative (DMSO) and positive (NEM) controls. WT CA II, WT CA III and mutant CA III proteins (C182S, C187S and CDS) (5 µM, 20 µL) were added to each assay plate well containing the selected compounds (4 equiv, 20 µM, 0.79% DMSO) for hit validation, counter-screening and modified cysteine mapping experiments. Samples were incubated in the dark at room temperature prior to nMS analysis. For WT CA III, nMS analysis was performed at 1, 4 and ~16 h; for other samples, nMS analysis was performed following ~16-h incubation.
For concurrent binding of active site noncovalent inhibitor and electrophilic fragments, hit 2 was plated in duplicate (40 nL, 10 mM) as representative of the top binding hit pharmacophore (backfilled with DMSO to 160-nL total volume) – with one replicate serving as controls for covalent only binding samples and the other replicate enabling covalent compound binding prior to adding the noncovalent binder acetazolamide, a strong affinity pan-CA inhibitor and noncovalent active site binder. NEM was used in parallel as the positive control (40 nL, 10 mM). Acetazolamide was plated (20 nL, 10 mM) in triplicate. One replicate was backfilled with DMSO to 160-nL total volume to give the acetazolamide only binding control. The other replicates were used in combination with respectively NEM and 2 for concurrent binding screening. Columns 1 and 2 were prepared similarly to the primary screen with negative (DMSO) and positive (NEM) controls. WT CA III, WT CA II and the three CA III mutant proteins (C182S, C187S and CDS) were added (20 µL, 5 µM) to both duplicate wells of the assay plate containing NEM and 2 (4 equiv, 20 µM, 0.79% DMSO) and incubated in the dark at room temperature for ~16 h. In parallel, the CA proteins (20 µL, 5 µM) were incubated with one replicate of acetazolamide (2 equiv, 10 µM, 0.79% DMSO) for 30 min. One replicate of the CA protein samples with NEM and 2 was then transferred to the remaining acetazolamide (2 equiv, 10 µM, 0.9% DMSO) wells and concurrently incubated for 30 min. nMS analyses were performed following completion of incubation(s).
Native mass spectrometry instrumentation and acquisition
Mass spectra were acquired on either a SolariX 12 Tesla Fourier Transform–Ion Cyclotron Resonance Mass Spectrometer (FT-ICR MS) (Bruker) or a Q-Exactive Ultra High Mass Range (UHMR) Hybrid Quadrupole-Orbitrap Mass Spectrometer (MS) (ThermoFisher Scientific). One replicate of primary screening was completed on each MS instrument and the other binding studies were completed as indicated in data tables or figure legends. For nMS analysis, 3 µL of each sample was infused into the instrument in positive ion mode using a TriVersa NanoMate nanoESI source (Advion Biosciences) with a chip containing 5-µm diameter nozzles for nanoESI. Nanomate parameters included a voltage of 1.7 kV and a pressure of 1 PSI (~6.9 kPa). Parameters for both MS instruments were optimised to maximise signal intensity and signal-to-noise ratio while ensuring retention of folded intact proteins. The MS instrument parameters for the FT-ICR MS were as previously reported by Yu et al.20; however, skimmer 1 was varied between 30 and 120 V to assess covalent (120 V) and noncovalent (30 V) binding, with fragment screening completed at 120 V. The MS parameters for the UHMR MS were as follows: resolution 200,000, microscans 5, averaging 5, S-lens RF level at 200, extended trapping 1.0 eV, desolvation voltage between −15 V (noncovalent binding) to −50 V (covalent binding) with screening completed at both −15 and −50 V, all other experiments completed at −15 V, trapping gas pressure setting 1.0, capillary temperature 275°C, detector m/z optimisation set for low m/z, ion transfer target m/z set for high m/z, nitrogen as the collision gas, scan range m/z 1500–6000, data acquisition length for at least 0.5–1 min.
Native mass spectrometry data analysis
Native mass spectrometry screening data were analysed directly (with Bruker Compass DataAnalysis 6.1 for the FT-ICR MS spectra and Thermo Scientific Freestyle 1.8 SP2 QF1 for the UHMR MS spectra). Data were also batch deconvoluted in UniDec (ver. 7.0.2, see https://github.com/michaelmarty/UniDec)48 and visualised as a heatmap using an inhouse Python script to assist rapid identification and quantitation of ligand binding (or hits) as previously reported.20 Data analysis parameters were as follows: FT-ICR MS data: UniDec parameters: m/z 2000–5000 range, 1–20 charge state range, 29,000–31,000-Da MW range, sample mass every 1.0 Da, peak detection range and threshold of 10 Da and 0.05; script parameters: 29,000–31,000-Da MW range. UHMR MS data: UniDec parameters: m/z 2000–6000 range, 7–11 charge state range, 29,000–30,500-Da MW range, sample mass every 1.0 Da, peak detection range and threshold of 100 Da and 0.05; script parameters: 29,000–31,000-Da MW range.
Supplementary material
Supporting material includes representative nMS spectra for each identified hit pharmacophore; specificity binding to map the cysteine binding site of top hits against WT and mutant CA III (C182S, C187S, CDS); specificity, promiscuity and cell viability data for 1–12. Supplementary material is available online.
Data availability
The data that support this study are available in the article and accompanying online supplementary material.
Declaration of funding
The authors gratefully acknowledge support of the Clive and Vera Ramaciotti Foundation (Biomedical Research Award, grant number 2023BRA19, to L. M. Sternicki and S.-A. Poulsen) and the Australian Government through an Australian Research Council’s Industrial Transformation Training Centre grant (grant number IC180100021, to S.-A. Poulsen); Linkage, Infrastructure, Equipment and Facilities grant (grant number LE120100170, to S.-A. Poulsen); and Discovery grant (grant number DP220102618, to S.-A. Poulsen). We thank Therapeutic Innovation Australia for support of this project through their project voucher scheme (to K. F. Tonissen and L. M. Sternicki). We thank Griffith University for a Postgraduate Scholarship awarded to Y. Yu.
Acknowledgements
We thank Dr Emily Furlong and Dr Sarah Mueller for helpful discussions. We thank Dr Wendy Loa for mass spectrometry instrumentation support. The authors acknowledge the facilities and the scientific and technical assistance of Compounds Australia and the Ramaciotti Australian Native Mass Spectrometry Platform for Health Discoveries, Griffith University. These facilities are supported by Therapeutic Innovation Australia (TIA). TIA is supported by the Australian Government through the National Collaborative Research Infrastructure Strategy (NCRIS) program.
References
1 Boike L, Henning NJ, Nomura DK. Advances in covalent drug discovery. Nat Rev Drug Discov 2022; 21(12): 881-898.
| Crossref | Google Scholar | PubMed |
2 Dalton SE, Di Pietro O, Hennessy E. A medicinal chemistry perspective on FDA-approved small molecule drugs with a covalent mechanism of action. J Med Chem 2025; 68(3): 2307-2313.
| Crossref | Google Scholar | PubMed |
3 Guo Y, shuai W, Tong A, Wang Y. Advanced technologies for screening and identifying covalent inhibitors. Trends Anal Chem 2024; 178: 117833.
| Crossref | Google Scholar |
4 Moore AR, Rosenberg SC, McCormick F, Malek S. RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discov 2020; 19(8): 533-552.
| Crossref | Google Scholar | PubMed |
5 Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019; 575(7781): 217-223.
| Crossref | Google Scholar | PubMed |
6 Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, Chen JJ, Chen N, Frohn MJ, Goodman G, et al. Discovery of a covalent inhibitor of KRASG12C (AMG 510) for the treatment of solid tumors. J Med Chem 2020; 63(1): 52-65.
| Crossref | Google Scholar | PubMed |
7 Mahran R, Kapp JN, Valtonen S, Champagne A, Ning J, Gillette W, Stephen AG, Hao F, Plückthun A, Härmä H, et al. Beyond KRAS(G12C): biochemical and computational characterization of sotorasib and adagrasib binding specificity and the critical role of H95 and Y96. ACS Chem Biol 2024; 19(10): 2152-2164.
| Crossref | Google Scholar | PubMed |
8 Dhillon S. Adagrasib: first approval. Drugs 2023; 83(3): 275-285.
| Crossref | Google Scholar | PubMed |
9 Hammond J, Leister-Tebbe H, Gardner A, Abreu P, Bao W, Wisemandle W, Baniecki M, Hendrick VM, Damle B, Simón-Campos A, et al. Oral nirmatrelvir for high-risk, nonhospitalized adults with COVID-19. N Engl J Med 2022; 386(15): 1397-1408.
| Crossref | Google Scholar | PubMed |
10 Blair HA. Ritlecitinib: first approval. Drugs 2023; 83(14): 1315-1321.
| Crossref | Google Scholar | PubMed |
11 Xu H, Jesson MI, Seneviratne UI, Lin TH, Sharif MN, Xue L, Nguyen C, Everley RA, Trujillo JI, Johnson DS, et al. PF-06651600, a dual JAK3/TEC family kinase inhibitor. ACS Chem Biol 2019; 14(6): 1235-1242.
| Crossref | Google Scholar | PubMed |
12 Xu W, Kang C. Fragment-based drug design: from then until now, and toward the future. J Med Chem 2025; 68(5): 5000-5004.
| Crossref | Google Scholar | PubMed |
13 Resnick E, Bradley A, Gan J, Douangamath A, Krojer T, Sethi R, Geurink PP, Aimon A, Amitai G, Bellini D, et al. Rapid covalent-probe discovery by electrophile-fragment screening. J Am Chem Soc 2019; 141(22): 8951-8968.
| Crossref | Google Scholar | PubMed |
14 Wang G, Seidler NJ, Röhm S, Pan Y, Liang XJ, Haarer L, Berger B-T, Sivashanmugam SA, Wydra VR, Forster M, et al. Probing the protein kinases′ cysteinome by covalent fragments. Angew Chem Int Ed 2025; 64(8): e202419736.
| Crossref | Google Scholar | PubMed |
15 Keeley A, Ábrányi-Balogh P, Keserű GM. Design and characterization of a heterocyclic electrophilic fragment library for the discovery of cysteine-targeted covalent inhibitors. MedChemComm 2019; 10(2): 263-267.
| Crossref | Google Scholar | PubMed |
16 Edwards AN, Hsu K-L. Emerging opportunities for intact and native protein analysis using chemical proteomics. Analytica Chimica Acta 2025; 1338: 343551.
| Crossref | Google Scholar | PubMed |
17 Sternicki LM, Poulsen SA. Fragment-based drug discovery campaigns guided by native mass spectrometry. RSC Med Chem 2024; 15(7): 2270-2285.
| Crossref | Google Scholar | PubMed |
18 Sternicki LM, Poulsen S-A. Toward routine utilisation of native mass spectrometry as an enabler of contemporary drug development. RSC Med Chem 2025; [Published online early, 30 July 2025].
| Crossref | Google Scholar |
19 Poulsen S-A. Fragment screening by native state mass spectrometry. Aust J Chem 2013; 66(12): 1495-1501.
| Crossref | Google Scholar |
20 Yu Y, Sternicki LM, Hilko DH, Jarrott RJ, Di Trapani G, Tonissen KF, Poulsen S-A. Investigating active site binding of ligands to high and low activity carbonic anhydrase enzymes using native mass spectrometry. J Med Chem 2024; 67(17): 15862-15872.
| Crossref | Google Scholar | PubMed |
21 Poulsen S-A, Davis RA, Keys TG. Screening a natural product-based combinatorial library using FTICR mass spectrometry. Bioorg & Med Chem 2006; 14(2): 510-515.
| Crossref | Google Scholar | PubMed |
22 Gavriilidou AFM, Holding FP, Coyle JE, Zenobi R. Application of native ESI-MS to characterize interactions between compounds derived from fragment-based discovery campaigns and two pharmaceutically relevant proteins. SLAS Discov 2018; 23(9): 951-959.
| Crossref | Google Scholar | PubMed |
23 Peschke M, Verkerk UH, Kebarle P. Features of the ESI mechanism that affect the observation of multiply charged noncovalent protein complexes and the determination of the association constant by the titration method. J Am Soc Mass Spectrom 2004; 15(10): 1424-1434.
| Crossref | Google Scholar | PubMed |
24 Mehmood S, Allison TM, Robinson CV. Mass spectrometry of protein complexes: from origins to applications. Annu Rev Phys Chem 2015; 66: 453-474.
| Crossref | Google Scholar | PubMed |
25 Kitova EN, El-Hawiet A, Schnier PD, Klassen JS. Reliable determinations of protein–ligand interactions by direct ESI-MS measurements. Are we there yet? J Am Soc Mass Spectrom 2012; 23(3): 431-441.
| Crossref | Google Scholar | PubMed |
26 Boeri Erba E, Barylyuk K, Yang Y, Zenobi R. Quantifying protein–protein interactions within noncovalent complexes using electrospray ionization mass spectrometry. Anal Chem 2011; 83(24): 9251-9259.
| Crossref | Google Scholar | PubMed |
27 Supuran CT. A simple yet multifaceted 90 years old, evergreen enzyme: Carbonic anhydrase, its inhibition and activation. Bioorg & Med Chem Lett 2023; 93: 129411.
| Crossref | Google Scholar | PubMed |
28 Nishimori I, Minakuchi T, Onishi S, Vullo D, Cecchi A, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: cloning, characterization, and inhibition studies of the cytosolic isozyme III with sulfonamides. Bioorg & Med Chem 2007; 15(23): 7229-7236.
| Crossref | Google Scholar | PubMed |
29 Silagi ES, Batista P, Shapiro IM, Risbud MV. Expression of carbonic anhydrase III, a nucleus pulposus phenotypic marker, is hypoxia-responsive and confers protection from oxidative stress-induced cell death. Scientific Reports 2018; 8(1): 4856.
| Crossref | Google Scholar | PubMed |
30 Chai YC, Jung CH, Lii CK, Ashraf SS, Hendrich S, Wolf B, Sies H, Thomas JA. Identification of an abundant S-thiolated rat liver protein as carbonic anhydrase III; characterization of S-thiolation and dethiolation reactions. Arch Biochem Biophys 1991; 284(2): 270-278.
| Crossref | Google Scholar | PubMed |
31 Tu C, Chen X, Ren X, LoGrasso PV, Jewell DA, Laipis PJ, Silverman DN. Interactions of active-site residues and catalytic activity of human carbonic anhydrase III. J Biol Chem 1994; 269(37): 23002-23006.
| Google Scholar | PubMed |
32 Pace NJ, Weerapana E. Diverse functional roles of reactive cysteines. ACS Chem Biol 2013; 8(2): 283-296.
| Crossref | Google Scholar | PubMed |
33 Go YM, Chandler JD, Jones DP. The cysteine proteome. Free Radic Biol Med 2015; 84: 227-245.
| Crossref | Google Scholar | PubMed |
34 Yu Y, Poulsen S-A, Di Trapani G, Tonissen KF. Exploring the redox and pH dimension of carbonic anhydrases in cancer: a focus on carbonic anhydrase 3. Antioxid Redox Signal 2024; 41(13–15): 957-975.
| Crossref | Google Scholar | PubMed |
35 Thomas RP, Heap RE, Zappacosta F, Grant EK, Pogány P, Besley S, Fallon DJ, Hann MM, House D, Tomkinson NCO, et al. A direct-to-biology high-throughput chemistry approach to reactive fragment screening. Chem Sci 2021; 12(36): 12098-12106.
| Crossref | Google Scholar |
36 Glöckner S, Heine A, Klebe G. A proof-of-concept fragment screening of a hit-validated 96-compounds library against human carbonic anhydrase II. Biomolecules 2020; 10(4): 518.
| Crossref | Google Scholar | PubMed |
37 Del Giudice R, Monti DM, Truppo E, Arciello A, Supuran CT, De Simone G, Monti SM. Human carbonic anhydrase VII protects cells from oxidative damage. Biol Chem 2013; 394(10): 1343-1348.
| Crossref | Google Scholar | PubMed |
38 Dubiella C, Pinch BJ, Koikawa K, Zaidman D, Poon E, Manz TD, Nabet B, He S, Resnick E, Rogel A, et al. Sulfopin is a covalent inhibitor of Pin1 that blocks Myc-driven tumors in vivo. Nat Chem Biol 2021; 17(9): 954-963.
| Crossref | Google Scholar | PubMed |
39 Ghebremariam YT, Erlanson DA, Cooke JP. A novel and potent inhibitor of dimethylarginine dimethylaminohydrolase: a modulator of cardiovascular nitric oxide. J Pharmacol Exp Ther 2014; 348(1): 69-76.
| Crossref | Google Scholar | PubMed |
40 Gehringer M, Laufer SA. Emerging and re-emerging warheads for targeted covalent inhibitors: applications in medicinal chemistry and chemical biology. J Med Chem 2019; 62(12): 5673-5724.
| Crossref | Google Scholar | PubMed |
41 Craven GB, Affron DP, Kösel T, Wong TLM, Jukes ZH, Liu CT, Morgan RML, Armstrong A, Mann DJ. Multiparameter kinetic analysis for covalent fragment optimization by using quantitative irreversible tethering (qIT). Chembiochem 2020; 21(23): 3417-3422.
| Crossref | Google Scholar | PubMed |
42 Shannon DA, Weerapana E. Covalent protein modification: the current landscape of residue-specific electrophiles. Curr Opin Chem Biol 2015; 24: 18-26.
| Crossref | Google Scholar | PubMed |
43 Enamine. Electrophilic Covalent Probe Library. Available at https://enamine.net/compound-libraries/covalent-libraries/electrophilic-covalent-probe-library [Verified 23 April 2025]
44 Moeker J, Mahon BP, Bornaghi LF, Vullo D, Supuran CT, McKenna R, Poulsen S-A. Structural insights into carbonic anhydrase IX isoform specificity of carbohydrate-based sulfamates. J Med Chem 2014; 57(20): 8635-8645.
| Crossref | Google Scholar | PubMed |
45 Mujumdar P, Teruya K, Tonissen KF, Vullo D, Supuran CT, Peat TS, Poulsen S-A. An unusual natural product primary sulfonamide: synthesis, carbonic anhydrase inhibition, and protein X-ray structures of psammaplin C. J Med Chem 2016; 59(11): 5462-5470.
| Crossref | Google Scholar | PubMed |
46 García-Nafría J, Watson JF, Greger IH. IVA cloning: a single-tube universal cloning system exploiting bacterial in vivo assembly. Scientific Reports 2016; 6(1): 27459.
| Crossref | Google Scholar | PubMed |
47 Simpson M, Poulsen S-A. An overview of Australia’s compound management facility: the Queensland Compound Library. ACS Chem Biol 2014; 9(1): 28-33.
| Crossref | Google Scholar | PubMed |
48 Marty MT. A universal score for deconvolution of intact protein and native electrospray mass spectra. Anal Chem 2020; 92(6): 4395-4401.
| Crossref | Google Scholar | PubMed |