

The Elusive Nitro-Functionalised Member of the IRMOF-9 Family
Luke Conte A , Tian-You Zhou B , Omid T. Qazvini B , Lujia Liu C , David R. Turner D , Shane G. Telfer B and Christopher Richardson A E
A E
A School of Chemistry and Biomolecular Science, Faculty of Science, Medicine and Health, University of Wollongong, Wollongong, NSW 2522, Australia.
B MacDiarmid Institute of Advanced Materials and Nanotechnology, Institute of Fundamental Sciences, Massey University, Palmerston North 4442, New Zealand.
C Department of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208-3113, USA.
D School of Chemistry, Monash University, Clayton, Vic. 3800, Australia.
E Corresponding author. Email: chris_richardson@uow.edu.au
Australian Journal of Chemistry 72(10) 811-816 https://doi.org/10.1071/CH19194
Submitted: 30 April 2019 Accepted: 4 July 2019 Published: 23 July 2019
Abstract
The solvothermal reaction of 2-nitro-[1,1′‐biphenyl]‐4,4′‐dicarboxylic acid (H2bpdcNO2) with Zn(NO3)2·6H2O in DMF solvent does not give a functionalised variant of IRMOF-9. Single-crystal X-ray diffraction analysis shows the major initial product of this reaction, WUF-21 (WUF = Wollongong University Framework), is a porous interpenetrated diamondoid metal–organic framework (MOF) with a secondary building unit that ‘doubly straps’ eight bridging bpdcNO2 ligands in a distorted tetrahedral shape around an unusual pentazinc core. A second porous MOF phase (WUF-23) containing a large and novel dodecazinc secondary building unit forms in the same reaction and eventually predominates in solutions containing formate anion, which arises from the hydrolysis of DMF. Doping the starting ligand with [1,1′‐biphenyl]‐4,4′‐dicarboxylic acid (H2bpdc) provides a facile way to grow nitro-functionalised IRMOF-9, hereafter denoted as WUF-22, where the dopant is carried through into the product. Activated WUF-22 is a microporous solid with an apparent Brunauer–Emmett–Teller (BET) surface area of 2497 m2 g−1, which matches well with geometric surface area calculations. The CO2 adsorption properties of WUF-22 are reported.
Introduction
We have long been interested in the functionalisation and post-synthetic modification of metal–organic frameworks (MOFs). Within this context, we have paid special attention to the interpenetrated primitive cubic (pcu) lattices that form from functionalised derivatives of linearly divergent [1,1′‐biphenyl]‐4,4′‐dicarboxylic acids and tetrazinc clusters.[1,2] The parent compound of this series was first described by Yaghi and coworkers as part of their demonstration of isoreticular framework expansion and was named IRMOF-9.[3] It is a true testament of the power of the isoreticular approach that over 30 functionalised IRMOF-9-type compounds have been described.[2c,4]
In the course of our studies, we had cause to prepare the nitro-functionalised IRMOF-9 starting from 2-nitro-[1,1′-biphenyl]-4,4′-dicarboxylic acid (H2bpdcNO2). Surprisingly, despite the popularity of H2bpdcNO2 as a linear bridging ligand for MOFs,[5] this IRMOF is unreported. We describe here our results of preparing this complex and show that doping provides a facile way of triggering crystal formation where standard methods fail.
Results and Discussion
Our standard solvothermal method of heating functionalised H2bpdc with a 3-fold excess of zinc nitrate hexahydrate at 100°C in DMF solvent overnight has been successful for preparing many IRMOF-9 complexes.[1a,1b,1d–g] However, application of this methodology to the reaction with H2bpdcNO2 failed to yield any crystals. Varying the reaction time, temperature, and concentration of the DMF solution also did not result in any crystal growth, much to our chagrin. The change in colour of the reaction solutions from colourless to a bright yellow indicated to us that some change had taken place, and several solutions were stored at room temperature. After a period of several months, three types of crystals began to deposit from the solutions. After 1 year, there were large yellow and large orange crystals and smaller colourless crystals (Figs S1, S2, Supplementary Material). Single crystal X-ray diffraction (SCXRD) on the orange crystals confirmed they were catena-(dimethylammonium tris(μ2-formato)zinc(ii) (Cambridge Structural Database (CSD) code DAXNIA01).[6] The large yellow crystals were agglomerates and could be cleaved to give smaller individual crystals. SCXRD analysis on these crystals and the smaller colourless rods (Fig. S2, Supplementary Material) revealed the structure crystallised in the monoclinic space group C2/c. The asymmetric unit contains two zinc–carboxylate clusters, each consisting of one-half Zn atom and two full Zn atoms with two bpdcNO2 linkers bridging the clusters. Each cluster then coordinates another bpdcNO2 linker (Fig. S3, Supplementary Material). The clusters are differentiated by Zn3 in one cluster binding two DMF ligands whereas Zn5 in the other cluster coordinates a DMF ligand and an aqua ligand. The remaining atom in each cluster is a µ3-hydroxido moiety and this participates in a H-bond with a non-coordinated carboxylate oxygen of a bridging bpdcNO2 linker (Fig. 1a). This gives a final formulation for this MOF of [Zn5(μ3-OH)2(bpdcNO2)4(DMF)3(H2O)], hereafter denoted as WUF-21. By virtue of Zn1 and Zn2 lying on 2-fold axes, the secondary building units (SBUs) are pentazinc clusters with the central Zn1 and Zn2 atoms in each cluster being six-coordinate and distorted octahedral in geometry. Fig. 1a shows the SBU based on Zn2, Zn3, and Zn6, and the hydrogen bonding between µ3-hydroxido and carboxylate groups. The coordination geometry of Zn3 may be considered distorted octahedral with a very long bond to a µ2-bridging carboxylate oxygen (Zn3⋯O20ii, 2.364(12) Å) and Zn6 is four-coordinate with tetrahedral geometry. The pentazinc SBUs coordinate eight bridging bpdcNO2 ligands in total, and these extend from the SBUs in pairs and in a distorted tetrahedral shape, making this a particularly bulky SBU (Fig. 1b). The combination of tetrahedral nodes and linear links reticulate into a diamondoid network (dia) with large pores. The pore space is partly occupied by an interpenetrating framework but there are pores in the structure parallel to the c axis, as shown in Fig. 1c. The porosity in the structure is supported by a void volume calculation in PLATON,[7] which returned a value of 49 % of the unit cell.

|
Another morphology of crystal generally prevailed with time. Synchrotron diffraction data revealed the complex crystallises in the space group P-1 with an asymmetric unit containing 12 Zn centres, 16 half-ligands of which 9 bear nitro groups, 4 bridging formates, 4 μ3-hydroxido linkages, 6 aqua ligands, 2 bound DMF, and a further 2 DMF solvate molecules, giving a formula of [Zn12(bpdcNO2)8(μ4-O2CH)4(μ3-OH)4(DMF)2(H2O)6·2DMF], named WUF-23. The half-ligands connect so that intact bpdcNO2 bridging ligands are formed with full-occupancy nitro groups. The zinc centres are assembled into a dodecazinc platter by bridging carboxylate and formate connectors. By virtue of the 16 half-ligands that project from the platter, not only does this SBU have high nuclearity but also high connectivity (Fig. S4, Supplementary Material). Eight octahedrally coordinated zinc centres run along a central vein, with the remaining four tetrahedrally coordinated centres around the platter’s periphery. The μ3-hydroxido moieties and the monodentate aqua ligands attached to Zn6 and Zn7 show stabilising intra-SBU H-bond interactions with carboxyl groups. Other zinc centres coordinate monodentate ligands: Zn1 and Zn12, which can be considered the terminal metal centres, each coordinate two aqua ligands, and Zn4 and Zn9 coordinate the DMF ligands.
The expanded structure is highly porous and a PLATON calculation returned a solvent accessible volume of 58 % of the unit cell. Alongside the high porosity is a tortuous pore system. Channels are evident parallel to each of the crystallographic axes (100, ~10 × 12 Å; 010, ~12 × 14 Å; 001, ~12 × 18 Å) as well as the 110 and 111 directions (Figs S5–S8, Supplementary Material). A view of the channel system permeating along the crystallographic 100 direction is shown in Fig. 2. The formation of this phase is dependent on sufficient formate being available as it plays a crucial role in the assembly of the SBU. The appearance of this phase and catena-(dimethylammonium tris(μ2-formato)zinc(ii) is consistent with the hydrolysis of DMF during solvothermal treatment and in the ageing solutions.
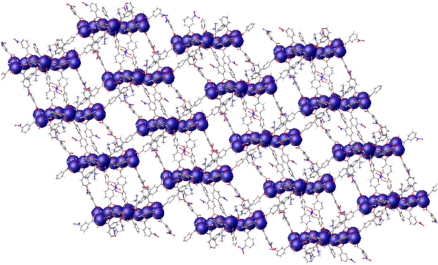
|
With our curiosity piqued by nitro-functionalised IRMOF-9 crystals failing to grow under our standard conditions, we hypothesised that doping H2bpdcNO2 with H2bpdc, the latter forming the IRMOF-9 structure, would solve this problem; our hypothesis being that H2bpdc would trigger crystal formation allowing the nitrated ligand to be incorporated in the growing lattice. In order to find the minimum amount of dopant needed, we spiked H2bpdcNO2 with small proportions of H2bpdc (4, 6, 9, and 12 mol-% of H2bpdc, as estimated by integration of 1H NMR spectra; Figs S12–S16, Supplementary Material), and reacted each mixture with three equivalents of zinc nitrate in DMF under identical conditions of solution concentration, time, and temperature. No crystal formation was observed with the reaction containing 4 mol-% doping and the WUF-21 structure formed after many months. Each of the other reactions gave clumps of crystals during heating overnight and the powder X-ray diffraction (PXRD) patterns of these samples showed good correlation to IRMOF-9 (Fig. 3; Figs S21–S23, Supplementary Material). This result clearly indicates a lower threshold exists for effective doping to direct phase selection in this system.
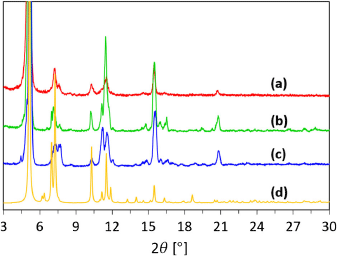
|
SCXRD analysis confirmed the crystals are nitro-functionalised analogues of IRMOF-9 and they were named WUF-22. The MOF crystallises as a pair of interpenetrating pcu networks in the space group C2/m. This is a space group we have found to be common for functionalised IRMOF-9 complexes.[1a,1b,1d,1f–h] The asymmetric unit features one full Zn atom (Zn2), two half Zn atoms (Zn1, Zn3), and a half oxido atom (O1) at the centre of the SBU, in addition to a quarter of a solvent water molecule occupying a special position (0.0, 0.5, 1). Zn3 coordinates a partially occupied water molecule (0.33). All atoms of one bpdcNO2 linker are contained in the asymmetric unit (O20 to O37), while the second ligand based on O2 to O16 has atoms C4, C5, C8, C11, C14, and C15 lying across a mirror plane (Fig. S9, Supplementary Material). The phenyl ring carbons C6, C7, C9, C10, and the nitro group attached to this ring (N90–O92) were assigned half occupancy to match the crystallographically defined disorder. Nitrogen atoms of the tag groups were found in difference maps and therefore define the positions of the nitro groups. These were completed in the crystallographic model by attaching oxygen atoms in fixed representative positions in the structure. Further details concerning the refinement can be found in the Supplementary Material.
The close association of the frameworks is sustained by short contacts (~2.6 Å) between C–H bonds of one framework to a carboxylate oxygen in the SBU of the partner framework (H7⋯O36, (1/2 − x, 1/2 + y, 1 − z)). This results in the largest pores in this structure running parallel to the crystallographic c axis, and lined with nitro groups (Fig. 4). The shortest atom centre-to-atom centre distances (i.e. excluding Van der Waals contact distances) in these pores range from 6 to 9 Å between nitro groups (Fig. S10, Supplementary Material) with distances of ~10.5 Å for most other interatom contacts around the pore environment.
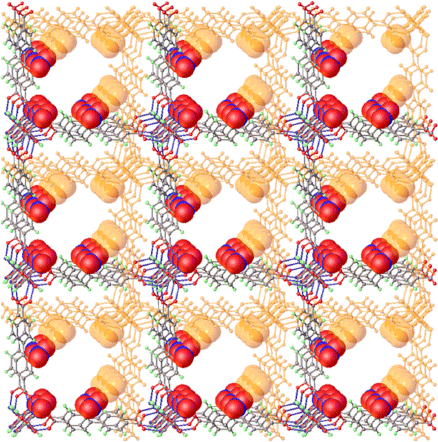
|
We determined the proportion of bpdc linkers in the resultant phases of WUF-22 by 1H NMR spectroscopy after digesting crystals in DCl/[D6]DMSO (Figs S17–S20, Supplementary Material). The results showed bpdc is present in higher amounts in the crystals than was supplied by the synthesis mixture at each doping level, pointing to a preferential incorporation of bpdc over bpdcNO2 (Table 1). The MOFs are named WUF-22(X) where X represents the percentage incorporation of bpdc. We have observed preferential incorporation of one ligand over another in multivariate IRMOF-1 compounds related to crystal growth rates[8] and in other IRMOF-9 compounds related to steric reasons.[1g] We postulate here that bpdc ligands nucleate crystal growth and continue to be incorporated into the structure, leading to their higher proportion.

|
Yields were recorded on samples after solvent exchange with CH2Cl2 and vacuum drying, and were found to depend strongly on the doping level (Table 1). The yield of 34 % obtained at the highest doping level of 12 mol-% of H2bpdc in H2bpdcNO2 is comparable with yields we have obtained in non-doped IRMOF-9 systems. As this doping level gave the highest yield, WUF-22(14) was used for all further analyses. In addition, in subsequent preparations, clumps of crystals formed by ~16 h were split apart with a spatula and this resulted in the growth of many small cubic-shaped crystals of WUF-22(14) over the next few hours.
Gas Adsorption Studies
PXRD patterns recorded after manual separation of the visibly discernible zinc formate crystals showed the remaining crystals were mixtures of WUF-21 and WUF-23, as expected (Fig. S24, Supplementary Material). Furthermore, solvent exchange of the mother liquor for fresh DMF resulted in visible degradation of the crystals. For these reasons, activation and gas sorption studies were not pursued.
Crystals of WUF-22(14) were used to study surface area and gas adsorption properties after solvent exchange with acetone, then cyclohexane, and activation by freeze-drying and heating under dynamic vacuum at 120°C. Simultaneous thermogravimetric analysis–differential scanning calorimetry (TG-DSC) on activated WUF-22(14) showed no thermal events in the sample until framework decomposition (Fig. S25, Supplementary Material). This contrasts with our work on a strontium-based MOF of this ligand (WUF-15) where exothermic loss of the nitro group was observed.[9] The N2 isotherm at 77 K is Type I (Fig. 5) and consistent with the expected microporosity of IRMOF-9-type compounds. The apparent BET surface area calculated in the range 0.008–0.04 P/P0 was 2497 m2 g−1 with a pore volume of 0.901 cm3 g−1 at 0.95 P/P0. Pore size analysis derived from density functional theory (DFT) calculations determined a tight distribution clustered around 10 Å (Fig. 5b) and this agrees well with the results from SCXRD.
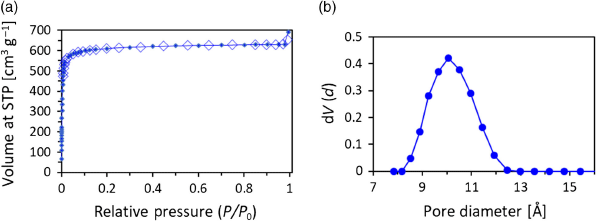
|
To support our experimental results, we performed surface area calculations using the method of Snurr with a probe atom radius set to 3.72 Å to match dinitrogen.[10] Ordered models containing four tetrazinc clusters and twelve unique ligands in the unit cell were constructed with formulas [(Zn4O)4(bpdc-NO2)11(bpdc)1] and [(Zn4O)4(bpdc-NO2)10(bpdc)2] to bound the formula of WUF-22(14) [(Zn4O)4(bpdc-NO2)10.44(bpdc)1.64]. The experimental result of 2497 m2 g−1 falls between the calculated values for [(Zn4O)4(bpdc-NO2)11(bpdc)1] at 2466 m2 g−1 and [(Zn4O)4(bpdc-NO2)10(bpdc)2] at 2510 m2 g−1, indicating a high-quality sample of WUF-22(14) with complete activation.
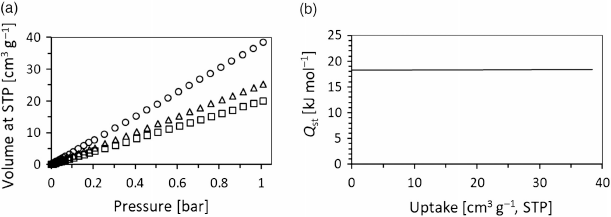
Given the nitro-functionalisation of WUF-22(14), we were in interested to see how it performed in the adsorption of CO2 and how this compared with other MOF materials.[4f,11] Fig. 6a shows the adsorption legs of CO2 isotherms collected at 273, 288, and 298 K up to 1 bar (100 kPa) with full adsorption–desorption isotherms provided in Fig. S27 (Supplementary Material). Each adsorption isotherm is linear and WUF-22(14) takes up a maximum of 38.5 cm3 g−1 at 1 bar and 273 K. In comparison with other functionalised IRMOF-9-type compounds from our laboratories, this value is surprisingly low. As examples, sulfone-functionalised IRMOF-9 frameworks had uptakes between 44 and 51 cm3 g−1 and the uptakes of dimethylthiocarbamate-functionalised IRMOF-9 complexes were 49–56 cm3 g−1 at 273 K. The poor adsorption of CO2 is responsible for a low selectivity factor over N2 of 6.7, as determined from single-component gas isotherms at 298 K and a hypothetical gas composition of 16 % CO2, 77 % N2, and 7 % from other gases (see Supplementary Material).
|
The isosteric heat of adsorption (Qst) for CO2 binding to WUF-22(14) was calculated from the adsorption isotherms collected at all three temperatures after fitting them with a virial equation (Fig. S28, Supplementary Material). This results in a Qst of approximately −18 kJ mol−1 across all uptake values (Fig. 6b). This modest value suggests that CO2 does not interact strongly with potential adsorption sites like the framework’s nitro groups. By way of comparison, the Qst for IRMOF-9 was calculated by a virial method to be approximately −23 kJ mol−1[4f] and the value for nitro-functionalised IRMOF-8 is approximately −35 kJ mol−1.[11a] We note that care needs to be taken in comparisons with other MOFs, particularly where the apparent BET surface areas fall well short of calculated surface areas and that to determine a true measure of the interaction of CO2 with organic functional groups in MOFs requires experimental surface areas close to those from simulation or calculation. For example, it is known that small amounts of remaining solvents in MOFs, particularly water, greatly impact the uptake and binding strength of the MOF towards CO2.
Conclusions
Two novel nitro-functionalised MOFs are the products after solvothermal reaction of pure H2bpdcNO2 with Zn(NO3)2·6H2O in DMF. We have shown that spiking ligands with proportions as low as 6 % can effectively direct the course of MOF formation towards desired products. The method is simple and capable of producing MOF materials that cannot be made under normal circumstances. Here, this enabled the preparation of a nitro-functionalised IRMOF-9 complex, which showed a surprisingly low affinity for CO2 after complete activation. We note the difficulty in making comparisons with other MOFs as experimental surface areas need to follow calculated values closely to ensure activation is achieved without some level of structural collapse or from frameworks with high levels of defects.
Experimental
All chemicals used were of analytical grade and purchased from Sigma–Aldrich, VWR Australia, or Ajax Finechem Pty Ltd. H2bpdcNO2 was prepared as described.[2b] 1H NMR spectra were obtained using a Bruker Ascend 400 MHz spectrometer and referenced to the residual protio peak at δ 2.50 ppm in [D6]DMSO. 1H NMR analysis was performed on MOF samples (~10 mg) digested by adding 35 % DCl in D2O (2.5 μL) and [D6]DMSO (500 μL) and stirring until a solution was obtained. Simultaneous TG-DSC data were obtained using a Netzsch STA 449 F1 Jupiter instrument. Measuring parameters of 10°C min−1 under a mix of nitrogen (10 cm3 min−1) and compressed air N2/O2 (80 : 20, 10 cm3 min−1) were used. PXRD patterns were recorded on a GBC-MMA X-ray diffractometer using Cu K(α) radiation (1.5418 Å) with samples mounted on 25-mm SiO2 substrates. The experimental data were collected in the 2θ angle range of 3–30° with a step size of 0.02° and a scan speed of 1° min−1.
SCXRD data for WUF-21 and WUF-22 were recorded on a Rigaku Spider diffractometer equipped with a MicroMax MM007 rotating anode generator (Cu radiation, 1.54180 Å), fitted with high-flux Osmic multilayer mirror optics, and a curved image-plate detector. Data were collected at 292 K and were integrated, scaled and averaged with FS Process.[12] XPREP was used to determine the space group and the structure was solved using SHELXT[13] for WUF-21 and SHELXS97[14] for WUF-22, and both were refined with SHELXL.[15] Data for WUF-23 were recorded on the MX2 beamline at the Australian Synchrotron with a wavelength of 0.71073 Å at 100 K. Two datasets were merged and truncated to 1.00 Å to give 97 % completeness. The structure was solved by direct methods and refined using SHELXL under the Olex2 graphical user interface.[16] Further details on the collections and refinements can be found in the Supplementary Material to this article.
Gas adsorption studies up to 1 bar were carried out using a Quantachrome Autosorb MP instrument and high-purity nitrogen (99.999 %) and carbon dioxide (99.995 %) gases at the Wollongong Isotope Geochronology Laboratory. Surface areas were determined using BET calculations. Pore size distributions were calculated using the quenched solid DFT equilibrium kernel for N2 at 77 K on carbon with slit or cylindrical pores as implemented in the Quantachrome software (v3.0).
Synthetic Procedure for WUF-21 and WUF-23
In a typical experiment, H2bpdcNO2 (105.6 mg, 0.37 mmol) and Zn(NO3)2·6H2O (365 mg, 1.22 mmol) were dissolved in DMF (10 mL) with stirring. The colourless solution was then placed in a preheated oven at temperatures from 80°C up to 120°C and for times between 24 and 48 h. No crystals formed in these brightly yellow coloured solutions. Samples were left to stand in capped vials at ambient temperature. Crystals started forming after approximately 6 months and some solutions were stored for over 3 years. No yields were calculated.
General Procedure for WUF-22(X) Synthesis
H2bpdc and H2bpdcNO2 in the desired molar ratio (50 mg in total combined mass) and Zn(NO3)2·6H2O (156 mg, 0.52 mmol) were dissolved in DMF (8 mL) with stirring. The solution was then placed in a preheated oven at 100°C for 18 h. For WUF-22(14), any clumps of crystals at this time were split apart using a spatula and heating was continued for another 6 h. The DMF solution was then exchanged three times with fresh DMF (2 mL) at 100°C and then with dry acetone at 80°C, each for 15 min. The crystals were left to stand under fresh dry acetone until needed for analysis, at which time the solvent was changed to cyclohexane at room temperature and the samples were activated by freeze-drying at −53°C and 0.09 mbar (9 Pa) for 1 h, followed by heating under dynamic vacuum at 120°C for 6 h. Details on yields and formulations are provided in Table S1 (Supplementary Material).
Crystallographic Data
Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre, CCDC deposition numbers 1912369, 1912370, and 1937365 for WUF-21–23, respectively. Copies of the data can be obtained free of charge from http://www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Centre (12 Union Road, Cambridge, CB2 1EZ, UK; fax: +44 1223 336033; email: deposit@ccdc.cam.ac.uk).
Supplementary Material
1H NMR spectra for the doped ratios of H2bpdc in H2bpdcNO2, digestion spectra for WUF-22(8–14), TG-DSC data for WUF-22(14), SCXRD data for WUF-21–23, PXRD patterns, adsorption–desorption gas sorption isotherms for WUF-22(14), and virial fitting plots and parameters are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
L.C. acknowledges the Australian Government for an Australian Government Research Training Program Award. C.R. thanks the University of Wollongong for financial support. This research was undertaken in part using the MX2 beamline at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector.[17] This research did not receive any specific funding.
References
[1] (a) A. D. Burrows, C. Frost, M. F. Mahon, C. Richardson, Angew. Chem. Int. Ed. 2008, 47, 8482.| Crossref | GoogleScholarGoogle Scholar |
(b) A. D. Burrows, C. G. Frost, M. F. Mahon, C. Richardson, Chem. Commun. 2009, 4218.
| Crossref | GoogleScholarGoogle Scholar |
(c) A. D. Burrows, L. C. Fisher, D. Hodgson, M. F. Mahon, N. F. Cessford, T. Dueren, C. Richardson, S. P. Rigby, CrystEngComm 2012, 14, 188.
| Crossref | GoogleScholarGoogle Scholar |
(d) A. D. Burrows, S. O. Hunter, M. F. Mahon, C. Richardson, Chem. Commun. 2013, 990.
| Crossref | GoogleScholarGoogle Scholar |
(e) M. R. Bryant, C. Richardson, CrystEngComm 2015, 17, 8858.
| Crossref | GoogleScholarGoogle Scholar |
(f) T. A. Ablott, M. Turzer, S. G. Telfer, C. Richardson, Cryst. Growth Des. 2016, 16, 7067.
| Crossref | GoogleScholarGoogle Scholar |
(g) M. R. Bryant, A. D. Burrows, C. J. Kepert, P. D. Southon, O. T. Qazvini, S. G. Telfer, C. Richardson, Cryst. Growth Des. 2017, 17, 2016.
| Crossref | GoogleScholarGoogle Scholar |
(h) M. R. Bryant, T. A. Ablott, S. G. Telfer, L. Liu, C. Richardson, CrystEngComm 2019, 21, 60.
| Crossref | GoogleScholarGoogle Scholar |
[2] (a) R. K. Deshpande, J. L. Minnaar, S. G. Telfer, Angew. Chem. Int. Ed. 2010, 49, 4598.
| Crossref | GoogleScholarGoogle Scholar |
(b) D. J. Lun, G. I. N. Waterhouse, S. G. Telfer, J. Am. Chem. Soc. 2011, 133, 5806.
| Crossref | GoogleScholarGoogle Scholar |
(c) R. K. Deshpande, G. I. Waterhouse, G. B. Jameson, S. G. Telfer, Chem. Commun. 2012, 1574.
| Crossref | GoogleScholarGoogle Scholar |
(d) A. Ferguson, L. Liu, S. J. Tapperwijn, D. Perl, F. X. Coudert, S. Van Cleuvenbergen, T. Verbiest, M. A. van der Veen, S. G. Telfer, Nat. Chem. 2016, 8, 250.
| Crossref | GoogleScholarGoogle Scholar |
[3] M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O’Keeffe, O. M. Yaghi, Science 2002, 295, 469.
| Crossref | GoogleScholarGoogle Scholar | 11799235PubMed |
[4] (a) T.-H. Park, K. Koh, A. G. Wong-Foy, A. J. Matzger, Cryst. Growth Des. 2011, 11, 2059.
| Crossref | GoogleScholarGoogle Scholar |
(b) J. M. Roberts, O. K. Farha, A. A. Sarjeant, J. T. Hupp, K. A. Scheidt, Cryst. Growth Des. 2011, 11, 4747.
| Crossref | GoogleScholarGoogle Scholar |
(c) D. Rankine, A. Avellaneda, M. R. Hill, C. J. Doonan, C. J. Sumby, Chem. Commun. 2012, 10328.
| Crossref | GoogleScholarGoogle Scholar |
(d) I. Boldog, L. Xing, A. Schulz, C. Janiak, C. R. Chim. 2012, 15, 866.
| Crossref | GoogleScholarGoogle Scholar |
(e) W. W. Lestari, P. Lönnecke, M. B. Sárosi, H. C. Streit, M. Adlung, C. Wickleder, M. Handke, W.-D. Einicke, R. Gläser, E. Hey-Hawkins, CrystEngComm 2013, 15, 3874.
| Crossref | GoogleScholarGoogle Scholar |
(f) R. Babarao, C. J. Coghlan, D. Rankine, W. M. Bloch, G. K. Gransbury, H. Sato, S. Kitagawa, C. J. Sumby, M. R. Hill, C. J. Doonan, Chem. Commun. 2014, 3238.
| Crossref | GoogleScholarGoogle Scholar |
(g) P. V. Dau, S. M. Cohen, Inorg. Chem. 2015, 54, 3134.
| Crossref | GoogleScholarGoogle Scholar |
(h) S. Glomb, D. Woschko, G. Makhloufi, C. Janiak, ACS Appl. Mater. Interfaces 2017, 9, 37419.
| Crossref | GoogleScholarGoogle Scholar |
[5] (a) P. V. Dau, S. M. Cohen, CrystEngComm 2013, 15, 9304.
| Crossref | GoogleScholarGoogle Scholar |
(b) L.-R. Guo, X.-L. Tang, Z.-H. Ju, K.-M. Zhang, H.-E. Jiang, W.-S. Liu, CrystEngComm 2013, 15, 9020.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. A. Gotthardt, S. Grosjean, T. S. Brunner, J. Kotzel, A. M. Ganzler, S. Wolf, S. Bräse, W. Kleist, Dalton Trans. 2015, 16802.
| Crossref | GoogleScholarGoogle Scholar |
(d) S. Halis, N. Reimer, A. Klinkebiel, U. Lüning, N. Stock, Microporous Mesoporous Mater. 2015, 216, 13.
| Crossref | GoogleScholarGoogle Scholar |
[6] P. Jain, N. S. Dalal, B. H. Toby, H. W. Kroto, A. K. Cheetham, J. Am. Chem. Soc. 2008, 130, 10450.
| Crossref | GoogleScholarGoogle Scholar | 18636729PubMed |
[7] A. L. Spek, Acta Crystallogr. Sect. D 2009, D65, 148.
[8] A. D. Burrows, L. C. Fisher, C. Richardson, S. P. Rigby, Chem. Commun. 2011, 47, 3380.
[9] A. Khansari, S. G. Telfer, C. Richardson, Cryst. Growth Des. 2019, 19, 268.
| Crossref | GoogleScholarGoogle Scholar |
[10] T. Düren, F. Millange, G. Férey, K. S. Walton, R. Q. Snurr, J. Phys. Chem. C 2007, 111, 15350.
| Crossref | GoogleScholarGoogle Scholar |
[11] (a) S. Orefuwa, E. Iriowen, H. Yang, B. Wakefield, A. Goudy, Microporous Mesoporous Mater. 2013, 177, 82.
| Crossref | GoogleScholarGoogle Scholar |
(b) D. C. Young, H. Yang, S. G. Telfer, P. E. Kruger, Inorg. Chem. 2017, 56, 12224.
| Crossref | GoogleScholarGoogle Scholar |
[12] FSProcess 1996 (Rigaku Corporation: Tokyo).
[13] G. M. Sheldrick, Acta Crystallogr. Sect. A 2015, A71, 3.
[14] G. M. Sheldrick, Acta Crystallogr. Sect. A 2008, A64, 112.
| Crossref | GoogleScholarGoogle Scholar |
[15] G. M. Sheldrick, Acta Crystallogr. Sect. C 2015, C71, 3.
[16] O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard, H. Puschmann, J. Appl. Cryst. 2009, 42, 339.
| Crossref | GoogleScholarGoogle Scholar |
[17] D. Aragao, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Rostan, R. Williamson, T. T. Caradoc-Davies, J. Synchrotron Radiat. 2018, 25, 885.
| Crossref | GoogleScholarGoogle Scholar | 29714201PubMed |