

Three Manganese Complexes of Anionic N4-Donor Schiff-Base Macrocycles: Monomeric MnII and MnIII, and dimeric MnIV*
Rajni K. Wilson A , Sébastien Dhers A , Stephen Sproules B , Eric J. L. McInnes C and Sally Brooker A D
A D
A Department of Chemistry and MacDiarmid Institute for Advanced Materials and Nanotechnology, University of Otago, PO Box 56, Dunedin 9054, New Zealand.
B WestCHEM, School of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK.
C School of Chemistry and Photon Science Institute, The University of Manchester, Oxford Road, Manchester M13 9PL, UK.
D Corresponding author. Email: sbrooker@chemistry.otago.ac.nz
Australian Journal of Chemistry 72(10) 805-810 https://doi.org/10.1071/CH19209
Submitted: 10 May 2019 Accepted: 25 June 2019 Published: 26 July 2019
Abstract
Three manganese macrocyclic complexes of two anionic N4-donor [1 + 1] Schiff-base macrocycles that differ in ring size (14 versus 16 membered), HLEt and HLPr (obtained from condensation of diphenylamine-2,2′-dicarboxaldehyde and either diethylenetriamine or dipropylenetriamine), are reported. Specifically, a pair of monomeric complexes MnIILEt(NCS)(H2O) and [MnIIILPr(NCS)2]·0.5H2O, plus a dimeric complex [MnIV2LEt2(O)2](ClO4)2·3DMF have been synthesised and characterised. Single crystal structure determinations on [MnIIILPr(NCS)2]·0.5H2O and [MnIV2LEt2(O)2](ClO4)2·3DMF revealed octahedral manganese centres in both cases: N6-coordinated Jahn–Teller distorted MnIII in the former and a pair of N4O2-coordinated MnIV in the latter. UV-Vis, IR, and electron paramagnetic resonance spectroscopy as well as magnetic measurements are reported. These macrocyclic complexes feature a simple and original design, and could find future uses as models for manganese catalase or as building blocks for the assembly of larger supramolecular architectures.
Introduction
Macrocycles are polydentate ligands with the donor atoms either incorporated in, or less commonly attached to, a cyclic backbone.[1] Porphyrins, chlorins, and corrins are classes of naturally occurring, highly conjugated, tetrapyrrolic, N4-donor macrocycles.[2–4] The complexes of these porphyrin-like N4-donor macrocycles are biologically important molecules, exhibiting a variety of critical functions, such as oxygen transport (e.g. haemoglobin, an iron complex of porphyrin), photosynthesis (e.g. chlorophylls, magnesium complexes of chlorins), and formation of blood in our body (e.g. vitamin B12, a cobalt complex of corrin).[3–5]
Manganese ions are involved in the active sites of several metalloenzymes that perform biological redox reactions.[3,6] In these natural systems, the manganese ions cycle through various different oxidation states during the catalytic cycle. Manganese catalases, which are responsible for the disproportionation of two molecules of hydrogen peroxide into water and oxygen, possess a dinuclear, O-bridged, manganese active site which cycles between MnII2 and MnII–MnIII during the catalytic cycle.[7,8] This, and interest in the Mn4Ca cluster in photosystem II which undergoes a cycle of four oxidation steps in order to catalyse the oxidation of two molecules of water into oxygen and protons,[9–17] has led to di- and polynuclear manganese complexes in a range of oxidation states being studied as models for the active sites of these systems.[18–26]
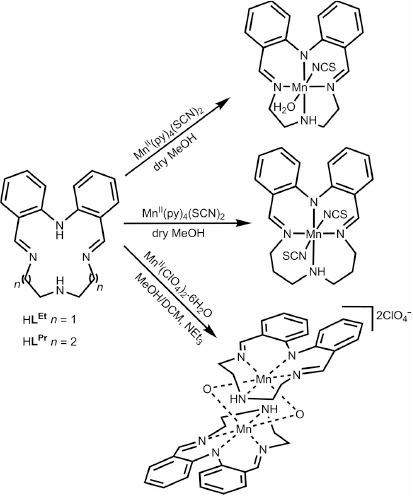
Macrocycles are a very useful ligand type for producing models of such biological active sites, not least as they provide a lot of control over the coordination site of the bound metal ion(s).[27] Interest in Schiff-base macrocycles in particular has remained high, and remains a particular strength in Australasia ever since the pioneering contributions to this field by Neil Curtis[28,29] and Richard Robson.[30] In this paper we describe the synthesis and characterisation of three manganese complexes of a pair of N4-donor [1 + 1] Schiff base macrocycles, differing only in ring size, developed previously in our group,[31–33] as we built on studies, published a decade earlier, by David Black.[34] While these anionic macrocycles, (LEt)− and (LPr)−, share some similarities with the naturally occurring porphyrin and porphyrin-like macrocycles (above), in that they all provide an N4-donor environment, herein they exhibit a significant difference: greater flexibility of coordination mode, as might be expected given the far lower extent of conjugation. The resulting manganese complexes, MnIILEt(NCS)(H2O), [MnIIILPr(NCS)2]·0.5H2O, and [MnIV2LEt2(O)2](ClO4)2·3DMF, vary in oxidation states (+II, +III, or +IV), ligand binding modes (meridional or facial), and nuclearity (monomeric or dimeric).
Results and Discussion
The pair of monomeric complexes, MnIILEt(NCS)(H2O) and [MnIIILPr(NCS)2]·0.5H2O, was synthesised in good yields (50 and 78 %) by the addition of one equivalent of MnII(py)4(SCN)2, under an inert atmosphere, to a suspension of the appropriate macrocycle in dry MeOH (Scheme 1). Upon addition of the metal salt the suspension immediately clarified and changed to a pale orange colour. Note that in both cases the pyridine present in the manganese thiocyanate reagent, MnII(py)4(SCN)2, is expected to have deprotonated the macrocycle.
|
After stirring under argon for 2 h, the colour slowly changed to dark brown for MnIILEt(NCS)(H2O). In the case of [MnIIILPr(NCS)2]·0.5H2O, the reaction solution was opened up to air after stirring for 4 h under Ar, which resulted in an immediate colour change from pale orange to dark brown. After exposure to air for 3 h with no obvious further colour change, the HLEt reaction solution was vapour diffused with diethyl ether, in air, giving MnIILEt(NCS)(H2O) as a brown solid. In the case of [MnIIILPr(NCS)2]·0.5H2O, a longer period of exposure to air, 7 days was found to be optimal to obtain the pure manganese(iii) complex, which conveniently precipitates out as a red solid. X-Ray quality dark red blocks of [MnIIILPr(NCS)2] were obtained by diethyl ether vapour diffusion into a MeOH solution of the reaction solution in air. An attempted synthesis of the manganese(iii) complex of the (LEt)− macrocycle resulted in the manganese(ii) complex.
The third complex was formed by complexation of HLEt with Mn(ClO4)2·6H2O and one equivalent of base (NEt3) in air, in a 1 : 1 mixture of dichloromethane and methanol, followed by diethyl ether vapour diffusion into the dark brown solution to give a dark brown solid. Diethyl ether vapour diffusion into a DMF solution of the sample, in air, gave a dark brown powder along with dark brown needles of [MnIV2LEt2(O)2](ClO4)2·3DMF suitable for an X-ray structure determination. The tentative assignment of the manganese(iv) oxidation state is based on the magnetic moment data and the structure determination (see below).
The positive ion electrospray mass spectra of MnIILEt(NCS)(H2O), [MnIIILPr(NCS)2]·0.5H2O, and the crystals of [MnIV2LEt2(O)2](ClO4)2·3DMF show a common peak, the fragment [Mn(LEt/Pr) – H]+, which is associated with the loss of an additional proton and the perchlorate or isothiocyanate co-ligand. Importantly, the MnIV2 dimer also shows a peak corresponding to a [Mn2LEt2(O)2(ClO4)]+ dimer fragment.
The infrared spectra of the three manganese complexes had imine stretches in the range 1611–1628 cm−1. In the (LEt)− complexes the imine stretches (1624 and 1628 cm−1) occurred at slightly lower energies than in the metal-free macrocycle HLEt (1629 cm−1) whereas in the (LPr)− complex the imine stretch (1611 cm−1) was at a significantly lower energy than in the crude metal-free HLPr macrocycle (1628 cm−1), as was observed in previously reported complexes of (LPr)−.[32] The ν(CN) stretch of the isothiocyanate ions in MnIILEt(NCS)(H2O) and [MnIIILPr(NCS)2]·0.5H2O is observed at 2050 and 2039 cm−1, respectively, and is consistent with a terminal N-bound ligand (literature range ≥ 2050 cm−1),[35] which was subsequently confirmed by X-ray crystallography (see below). The MnIV dimer showed two bands at 1074 and 621 cm−1, confirming the presence of the perchlorate anion.
The room temperature magnetic moment of 6.07 μB MnIILEt(NCS)(H2O) confirms a high-spin MnII S = 5/2 central ion. Its fluid solution electron paramagnetic resonance (EPR) spectrum recorded at X-band exhibited the characteristic six-lines from coupling of the electron spin to the 55Mn (I = 5/2, 100 % natural abundance) nucleus (Fig. S1, Supplementary Material). Simulation yielded giso = 2.0026 and a hyperfine coupling constant of Aiso = 89 × 10−4 cm−1 typical for MnII. The MnIII ion in [MnIIILPr(NCS)2]·0.5H2O is high-spin as denoted by its temperature independent μeff value of 4.91 μB (50–300 K) for an S = 2 ground state (Fig. S2, Supplementary Material). Below 50 K, μeff decreases due to the influence of zero-field splitting. The susceptibility has been successfully modelled with a sizeable |D| = 6.5 cm−1 at near full rhombicity, E/D = 0.029. These parameters are consistent with MnIII S = 2 in such a ligand field, and with being EPR silent at X-band.[36–38] A full variable temperature (VT) measurement conducted in the temperature range 2–300 K for [MnIV2LEt2(O)2](ClO4)2·3DMF (Fig. S3, Supplementary Material) gave a µeff value of 3.91 μB per Mn, consistent with 3 upe and hence MnIV. This µeff value drops only very slowly as the temperature is reduced, to a value of 2.74 μB, before dropping rapidly thereafter. This profile is consistent with weak antiferromagnetic coupling.
X-Ray Crystal Structures
Crystal structure determinations were carried out on [MnIIILPr(NCS)2] and [MnIV2LEt2(O)2](ClO4)2·3DMF. Examination of these structures revealed that [MnIIILPr(NCS)2] is mononuclear with the manganese(iii) ion in a Jahn–Teller distorted octahedral N6-donor environment whereas [MnIV2LEt2(O)2](ClO4)2·3DMF is dinuclear with the manganese(iv) ions also in an octahedral geometry.
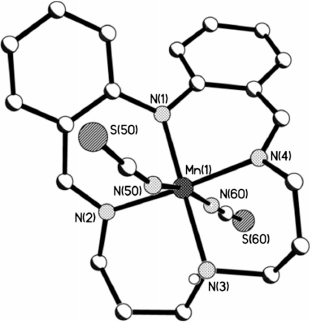
Dark red blocks of [MnIIILPr(NCS)2] were obtained by diethyl ether vapour diffusion into a MeOH solution of the sample and the crystal structure determined (Fig. 1).
|
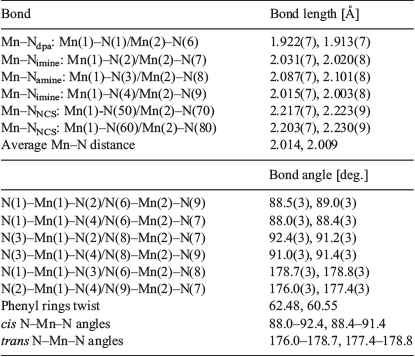
The complex crystallises in the monoclinic space group P21/n. The monoanionic tetradentate macrocyclic ligand is bound to the equatorial plane of the metal centre through all four nitrogen atoms (one amine, one deprotonated amine, and two imine nitrogen donors). In addition to the macrocycle, two axially coordinated (trans) isothiocyanate anions are also bound to the manganese(iii) centre, forming an N6 coordination environment. There are two entire [MnIIILPr(NCS)2] complexes in the asymmetric unit. The Mn(2) complex has no disorder whereas in the Mn(1) complex both CS atoms of the NCS− anions and the four alkyl carbon atoms adjacent to the N(3) atom and the NH proton in the central part of the dipropylene triamine alkyl chain are disordered over two sites (occupancy of 0.75 : 0.25).
The shortest Mn–N bond distance involves the deprotonated diphenylamine nitrogen atom, Mn–Ndpa (1.922(7) and 1.913(7) Å), whereas the longest is the Mn–Namine bond (2.087(7) and 2.101(8) Å). The four crystallographically independent Mn–Nimine bond lengths are equivalent within experimental error and are of intermediate lengths (average 2.017 Å; Table 1).
|
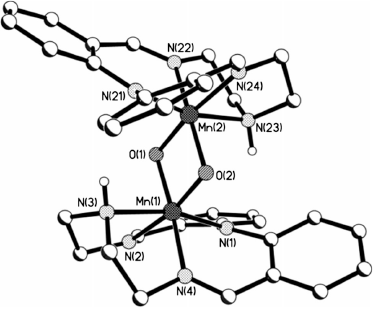
A few dark brown needles of [MnIV2LEt2(O)2](ClO4)2·3DMF were obtained by diethyl ether vapour diffusion into a DMF solution of the sample, in air, and the crystal structure determined (Fig. 2 and Table 2). The complex crystallises in the monoclinic space group P21/n with the entire complex in the asymmetric unit.
|

|
The geometry around each manganese centre is approximately octahedral, the ligating atoms being two cis oxo bridges and the four nitrogen atoms (one amine, one deprotonated amine, and two imine nitrogen donors) from the (LEt)− macrocycle. This is the first observation of this tetradentate macrocycle binding in a capping, not equatorial, mode (and hence the co-ligands occupying cis sites): all previous structurally characterised complexes feature an equatorial binding mode (with the co-ligands in trans sites). This 14-membered anionic N4-donor macrocycle has similarities to the 16-membered porphyrins, but this binding mode highlights a key difference: significantly greater flexibility (due to much less conjugation) which enables the capping binding mode observed here.
The assignment of the single atom bridges as µ-oxo rather than µ-hydroxo, hence also the manganese oxidation states as +IV not +III, was made only after careful consideration of the manganese coordination sphere, hydrogen bonding to another molecule, and CSD searches. No evidence of a proton was found near either oxo atom in the difference maps.
To further probe the assignment of manganese oxidation states in [MnIV2LEt2(O)2](ClO4)2·3DMF, a comparison with related structures was conducted. A search of the CSD (version 5.39) for N4-coordinated, bis-O (any type) bridged, dimanganese complexes resulted in 104 hits, of which only 74 involved bis-oxo bridged complexes (the others mostly involved phenolate O bridging). Narrowing the search to those structures where the oxidation states are clearly entered in the CSD as ‘manganese(X)’ where X is II, III, or IV, the manganese oxidation state combinations identified include 10 examples of MnIII2, 27 of MnIIIMnIV, and 5[39–43] of MnIV.
The average Mn–O (1.815(3) and 1.819(2) Å) and Mn–N distances (2.017 and 2.009 Å) in [MnIV2LEt2(O)2](ClO4)2·3DMF fall in the range for MnIV–O (1.772–1.829 (1.806) Å) and at the lower end of the range for MnIV–N (1.995–2.174 (2.077) Å) for literature bis(µ-oxo)-MnIV2 complexes. The Mn–O–Mn angles (96.8(1)° and 96.0(1)°) also fall in the range observed for related bis-oxo-MnIV2 complexes (95.1°–101.4° (98.01°)), but not in the range for bis-oxo-MnIII2 complexes (92.1°–94.8° (93.5°)). The Mn⋯Mn separation of 2.708(10) Å falls within the literature range for bis(µ-oxo)MnIV2 complexes (2.671–2.748 (2.724) Å), but also falls close to those reported for bis(µ-oxo)MnIII2 complexes (2.676–2.699 (2.689) Å).
A more detailed comparison of some of the key coordination sphere parameters in the present complex with those of three typical examples of structures selected from the CSD hits, in each of the three possible oxidation state combinations, MnIII2, localised MnIIIMnIV, and MnIV2, is presented in Table 3.[39,44,45] For ease of comparison we have defined the equatorial plane as comprising both oxo groups and the two nitrogen donors in the same plane (N2, N4; N22, N24; Fig. 2), with the axial direction comprising the remaining two nitrogen donors (N1, N3; N21, N23; Fig. 2). In [MnIV2LEt2(O)2](ClO4)2·3DMF the average axial Mn–N bond length (2.022 Å) is only slightly longer than the average equatorial bond length (2.004 Å), and both values are comparable to those in the bis(µ-oxo)dimanganese(iv) complex presented in Table 3, consistent with the oxidation state being MnIV2. No Jahn–Teller distortion is observed, unlike in the MnIII2 complex bis(µ-oxo)bis[N,N′-bis((6-methylpyrid-2-yl)methyl)-ethane-1,2-diamine]dimanganese(iii) perchlorate (Table 3),[44] so it is unlikely to be a MnIII2 or a localised MnIIIMnIV complex. The Mn–N distances also indicate that the bridging oxo groups do not induce a trans effect on the equatorial nitrogen atoms, something which has been seen in some other bis(µ-oxo)-MnIV2 complexes with Mn–Nequatorial >> Mn–Naxial.

|
In summary, the comparisons of Mn⋯Mn separation, average Mn–O and Mn–N distances, and Mn–O–Mn angles in [MnIV2LEt2(O)2](ClO4)2·3DMF with other complexes (Table 3) do not allow a definitive assignment of oxidation state, but overall it is most likely to be in the MnIV2 oxidation state.
Conclusions
Three manganese complexes, MnIILEt(NCS)(H2O), [MnIIILPr(NCS)2]·0.5H2O, and [MnIV2LEt2(O)2](ClO4)2·3DMF were prepared by deprotonation and metallation of the respective metal-free Schiff-base macrocycles, HLEt and crude HLPr. These anionic N4-donor macrocycles have similarities with the anionic N4-donor porphyrins, but also significant differences: herein their far greater flexibility, due to a much lower degree of conjugation, is demonstrated in the binding modes adopted: either meridional (equatorial square plane; trans co-ligands), or facial (capping; cis co-ligands). Also reported here are the first examples of these diphenylamine-2,2′-dicarboxaldehyde-based macrocycles facilitating access to 3d complexes in a wider range of oxidation states, +II, +III, and +IV: all previous complexes having been isolated only in the +II oxidation state. The mononuclear complexes produced are of interest as building blocks for larger supramolecular architectures. Our current efforts are focussed on refining the macrocycle design further, in particular aiming to increase the denticity and to include surface-anchoring groups.
Experimental
Magnetic data for MnIILEt(NCS)(H2O) was recorded at 300 K using a Quantum Design Physical Property Measurement System (PPMS) equipped with a vibrating sample mount with an applied field of 1 T. Variable temperature (2–300 K) magnetic susceptibility data for [MnIIILPr(NCS)2]·0.5H2O were recorded in a 1 T magnetic field on a SQUID magnetometer (MPMS Quantum Design). All magnetic data were corrected for diamagnetic contributions using Pascal’s constants. X-Band EPR data were collected on a Bruker EMX Micro spectrometer equipped with a ‘premium-X’ microwave bridge and simulated using Xsophe.[46] Elemental analyses were carried out by the Campbell Microanalytical Laboratory at the University of Otago.
MeOH and DMF were HPLC grade, whereas DCM was reagent grade and used without purification. MnII(py)4(SCN)2[35] and the HLEt macrocycle[31] were prepared as previously reported, but in the case of the HLPr macrocycle, rather than using the literature synthesis,[32] a crude sample of macrocycle was instead prepared without added acid (details below) and utilised without further purification.
MnIILEt(NCS)(H2O)
Under an Ar atmosphere, to a bright yellow suspension of the macrocycle HLEt (45.1 mg, 0.154 mmol) in methanol (30 mL) was added MnII(py)4(SCN)2 (75.3 mg, 0.154 mmol) as a solid, resulting in an pale orange coloured solution. After stirring under Ar for 2 h, the colour slowly changed to dark brown. The solution was stirred at room temperature overnight and then exposed to air for 7 h. The dark brown solution was concentrated to 20 mL under reduced pressure and finally subjected to diethyl ether vapour diffusion in air. The resulting brown solid was collected by filtration and dried under vacuum (33 mg, 50 %). Found: C 53.57, H 4.76, N 16.33, S 8.02. [C19H19N5SMn·H2O] (422.41 g mol−1) requires C 54.03, H 5.01, N 16.58, S 7.59 %. νmax (ATR)/cm−1 3061, 2864, 2050, 1624, 1598, 1527, 1483, 1454, 1434, 1398, 1289, 1194, 1156, 1039, 972, 857, 750, 622, 535, 473. m/z (ESI, MeCN) 345.0906; calcd for [MnIILEt – H]+ 345.0892. µeff (PPMS at 300 K) = 6.07 μB. λmax (DMF)/nm (ϵ/dm3 mol−1 cm−1) 343 (56904), 454 (2428). Λm (MeCN) = 53 Ω−1 cm2 mol−1.
[MnIIILPr(NCS)2]·0.5H2O
Under an Ar atmosphere, to a bright yellow suspension of the crude macrocycle HLPr (151.4 mg, 0.4725 mmol) in MeOH (40 mL) was added MnII(py)4(SCN)2 (230 mg, 0.4723 mmol) as a solid, causing an immediate colour change to pale orange. The solution was stirred under Ar for 4 h and then exposed to air which caused an immediate colour change to dark brown. After exposure to air for 7 days a dark red-brown precipitate was observed. The resulting dark red-brown solid was collected by filtration and dried under vacuum (185 mg, 78 %). Found: C 53.18, H 4.76, N 16.73, S 12.42. [C22H23N6S2Mn·0.5H2O] (499.53 g mol−1) requires C 52.90, H 4.84, N 16.82, S 12.84 %. νmax (ATR)/cm−1 3393, 3235, 2948, 2039, 1611, 1555, 1541, 1456, 1434, 1399, 1353, 1298, 1248, 1229, 1204, 1156, 1139, 1127, 1110, 1087, 1071, 1057, 1039, 989, 976, 954, 940, 886, 857, 782, 756, 734, 641, 606, 591, 552, 480, 468, 448, 434, 409. m/z (ESI+, MeCN) 373.1219; calcd for [MnIIILPr – H]+ 373.1185. µeff (SQUID at 300 K) = 4.91 μB. λmax (MeCN)/nm (ϵ/dm3 mol−1 cm−1) 315 (10884), 478 (5093), 550 (1472), 872 (309). Λm (MeCN) = 36 Ω−1 cm2 mol−1. X-Ray quality crystals were grown by slow evaporation of the reaction solution, giving red blocks of [MnIIILPr(NCS)2].
[MnIV2LEt2(O)2](ClO4)2·3.6H2O
In air, to a bright yellow solution of HLEt (90.4 mg, 0.309 mmol) in a 1 : 1 mixture of methanol and dichloromethane (40 mL) was added manganese(ii) perchlorate hexahydrate (112.0 mg, 0.309 mmol) as a solution in methanol (3 mL) resulting in a dark brown solution. A solution of triethylamine (31.1 mg, 0.309 mmol) was added resulting in no further colour change. This dark brown solution was stirred at room temperature overnight and concentrated to 20 mL by blowing compressed air over it and subjected to diethyl ether vapour diffusion. The dark brown solid was collected, dried under vacuum, and then left in air (65 mg, 21 %). Found: C 44.12, H 4.24, N 10.95. [C36H38N8Cl2O10Mn2·3.6H2O (988.38 g mol−1) requires C 43.75, H 4.61, N 11.34 %. νmax (ATR)/cm−1 3504, 3293, 2946, 1628, 1596, 1559, 1541, 1458, 1436, 1395, 1281, 1195, 1163, 1084, 972, 912, 875, 852, 810, 755, 649, 621, 593, 562, 538, 492, 457. m/z (ESI+, MeCN) 345.0906; calcd for [MnIILEt – H]+ 345.0895: 823.1358; calcd for [MnIV2LEt2(O)2(ClO4)]+ 823.1349. µeff (PPMS at 300 K) = 3.91 μB. λmax (MeCN)/nm (ϵ/dm3 mol−1 cm−1) 374 (8222), 485 (3529). A few single crystals, suitable for an X-ray structure determination, were obtained by diethyl ether vapour diffusion into a DMF solution of the sample, giving dark brown needles of [MnIV2LEt2(O)2](ClO4)2·3DMF.
X-Ray Crystallography
Data were collected on a Bruker Kappa Apex II area detector diffractometer at 89–90 K using graphite monochromated Mo-Kα radiation (λ 0.71073 Å). Both datasets (Tables S1 and S2, Supplementary Material) were absorption corrected using SCALE. The structures were solved using SHELXS-97 and refined against F2 using all data by full-matrix least-squares techniques with SHELXL-97. All non-hydrogen atoms were modelled anisotropically except where noted. Hydrogen atoms were inserted at calculated positions and rode on the atoms to which they were attached, except where noted. Crystallographic data has been deposited with the Cambridge Crystallographic Data Centre (reference numbers CCDC 1913017–1913018).
[MnIIILPr(NCS)2]
The asymmetric unit contains two [MnIIILPr(NCS)2] complexes. The Mn(2) complex has no disorder, whereas in the Mn(1) complex both the thiocyanate anions have the CS atoms disordered over two sites with 0.75 : 0.25 occupancy while the N atom is full occupancy. The alkyl carbon atoms adjacent to the N atoms in the dipropylenetriamine chain and the NH proton are disordered with 0.75 : 0.25 occupancy. The NH and the central C in each propylene chain are full occupancy. All non-hydrogen atoms were refined anisotropically except for the 0.75 and 0.25 occupancy C and N atoms of the dipropylenetriamine chain and the 0.75 and 0.25 occupancy C and S atoms of the thiocyanate anion which were left isotropic.
[MnIV2LEt2(O)2](ClO4)2·3DMF
The asymmetric unit comprised the entire macrocyclic dimeric complex, two perchlorates anions, and three DMF molecules of solvation. No disorder was present in the complex. One of the perchlorate anions was disordered over two sites with 0.8 : 0.2 occupancy. One of the DMF molecules was disordered over two sites with 0.7 : 0.3 occupancy. All non-hydrogen atoms were refined anisotropically except for the 0.2 occupancy perchlorate anion atoms, Cl25, O25, O26, O27, and O28. The 0.3 occupancy DMF atoms, C75, O75, N75, C76, and C77 were also refined isotropically.
Supplementary Material
Crystal data and structure refinement tables, EPR spectrum, and variable temperature magnetic moment figures are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by grants from the University of Otago (including a PhD scholarship for RKW) and the MacDiarmid Institute for Advanced Materials and Nanotechnology (including a PhD scholarship for SD). The authors are grateful for access to the EPSRC UK National EPR Facility.
References
[1] L. F. Lindoy, The Chemistry of Macrocyclic Ligand Complexes 1990 (Cambridge University Press: Cambridge).[2] N. V. Gerbeleu, V. B. Arion, J. Burgess, Template Synthesis of Macrocyclic Compounds 1999 (Wiley-VCH: Weinheim).
[3] A. Messerschmidt, R. Huber, T. Poulos, K. Wieghardt, Handbook of Metalloproteins, Vol. 1 2001 (John Wiley and Sons, Ltd: Chichester).
[4] K. M. Smith, Porphyrins and Metalloporphyrins 1976 (Elsevier/North-Holland Biomedical Press: New York, NY).
[5] A. I. Scott, Acc. Chem. Res. 1978, 11, 29.
| Crossref | GoogleScholarGoogle Scholar |
[6] See pp. 1–28 in: E. J. Larson, V. L. Pecoraro, Manganese Redox Enzymes 1992 (VCH: New York, NY).
[7] V. V. Barynin, M. M. Whittaker, S. V. Antonyuk, V. S. Lamzin, P. M. Harrison, P. J. Artymiuk, J. W. Whittaker, Structure 2001, 9, 725.
| Crossref | GoogleScholarGoogle Scholar | 11587647PubMed |
[8] N. N. Gerasimchuk, A. Gerges, T. Clifford, A. Danby, K. Bowman-James, Inorg. Chem. 1999, 38, 5633.
| Crossref | GoogleScholarGoogle Scholar | 11671294PubMed |
[9] U. F. Lingappa, D. R. Monteverde, J. S. Magyar, J. S. Valentine, W. W. Fischer, Free Radic. Biol. Med. 2019, in press.
| 30738765PubMed |
[10] R. O. Costa, S. S. Ferreira, C. A. Pereira, J. R. Harmer, C. J. Noble, G. Schenk, R. W. A. Franco, J. A. L. C. Resende, P. Comba, A. E. Roberts, C. Fernandes, A. Horn, Front Chem. 2018, 6, 491.
| 30456211PubMed |
[11] R. D. Britt, D. L. M. Suess, T. A. Stich, Proc. Natl. Acad. Sci. USA 2015, 112, 5265.
| Crossref | GoogleScholarGoogle Scholar | 25883270PubMed |
[12] J. Yano, V. Yachandra, Chem. Rev. 2014, 114, 4175.
| Crossref | GoogleScholarGoogle Scholar | 24684576PubMed |
[13] E. Y. Tsui, J. S. Kanady, T. Agapie, Inorg. Chem. 2013, 52, 13833.
| Crossref | GoogleScholarGoogle Scholar | 24328344PubMed |
[14] G. N. Ledesma, H. Eury, E. Anxolabéhère-Mallart, C. Hureau, S. R. Signorella, J. Inorg. Biochem. 2015, 146, 69.
| Crossref | GoogleScholarGoogle Scholar | 25771435PubMed |
[15] W.-T. Lee, S. B. Muñoz, D. A. Dickie, J. M. Smith, Angew. Chem. Int. Ed. 2014, 53, 9856.
| Crossref | GoogleScholarGoogle Scholar |
[16] K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber, S. Iwata, Science 2004, 303, 1831.
| Crossref | GoogleScholarGoogle Scholar | 14764885PubMed |
[17] S. Mukhopadhyay, S. K. Mandal, S. Bhaduri, W. H. Armstrong, Chem. Rev. 2004, 104, 3981.
| Crossref | GoogleScholarGoogle Scholar | 15352784PubMed |
[18] J. Glerup, P. A. Goodson, A. Hazell, R. Hazell, D. J. Hodgson, C. J. McKenzie, K. Michelsen, U. Rychlewska, H. Toftlund, Inorg. Chem. 1994, 33, 4105.
| Crossref | GoogleScholarGoogle Scholar |
[19] C. Philouze, G. Blondin, J.-J. Girerd, J. Guilhem, C. Pascard, D. Lexa, J. Am. Chem. Soc. 1994, 116, 8557.
| Crossref | GoogleScholarGoogle Scholar |
[20] S. Pal, W. H. Armstrong, Inorg. Chem. 1992, 31, 5417.
| Crossref | GoogleScholarGoogle Scholar |
[21] S. Mukhopadhyay, R. J. Staples, W. H. Armstrong, Chem. Commun. 2002, 864.
| Crossref | GoogleScholarGoogle Scholar |
[22] E. S. Koumousi, S. Mukherjee, C. M. Beavers, S. J. Teat, G. Christou, T. C. Stamatatos, Chem. Commun. 2011, 47, 11128.
| Crossref | GoogleScholarGoogle Scholar |
[23] R. K. Hocking, R. Brimblecombe, L.-Y. Chang, A. Singh, M. H. Cheah, C. Glover, W. H. Casey, L. Spiccia, Nat. Chem. 2011, 3, 461.
| Crossref | GoogleScholarGoogle Scholar | 21602861PubMed |
[24] M. Yagi, M. Toda, S. Yamada, H. Yamazaki, Chem. Commun. 2010, 46, 8594.
| Crossref | GoogleScholarGoogle Scholar |
[25] S. Brooker, V. McKee, W. B. Shepard, L. K. Pannell, J. Chem. Soc., Dalton Trans. 1987, 2555.
| Crossref | GoogleScholarGoogle Scholar |
[26] K. Wieghardt, U. Bossek, W. Gebert, Angew. Chem. Int. Ed. Engl. 1983, 22, 328.
| Crossref | GoogleScholarGoogle Scholar |
[27] V. McKee, Adv. Inorg. Chem. 1993, 40, 323.
[28] N. F. Curtis, D. A. House, Chem. Ind. 1961, 1708.
[29] N. F. Curtis, Supramol. Chem. 2012, 24, 439.
| Crossref | GoogleScholarGoogle Scholar |
[30] N. H. Pilkington, R. Robson, Aust. J. Chem. 1970, 23, 2225.
[31] R. Sanyal, S. A. Cameron, S. Brooker, Dalton Trans. 2011, 40, 12277.
| Crossref | GoogleScholarGoogle Scholar | 21909566PubMed |
[32] R. K. Wilson, S. Brooker, Dalton Trans. 2013, 42, 7913.
| Crossref | GoogleScholarGoogle Scholar | 23400271PubMed |
[33] R. K. Wilson, S. Brooker, Dalton Trans. 2013, 42, 12075.
| Crossref | GoogleScholarGoogle Scholar | 23860587PubMed |
[34] D. S. C. Black, N. E. Rothnie, Aust. J. Chem. 1983, 36, 2395.
| Crossref | GoogleScholarGoogle Scholar |
[35] J. G. Małecki, B. Machura, A. Świtlicka, T. Groń, M. Bałanda, Polyhedron 2011, 30, 746.
| Crossref | GoogleScholarGoogle Scholar |
[36] J. Krzystek, J. Telser, L. A. Pardi, D. P. Goldberg, B. M. Hoffman, L.-C. Brunel, Inorg. Chem. 1999, 38, 6121.
| Crossref | GoogleScholarGoogle Scholar | 11671322PubMed |
[37] G. Aromí, J. Telser, A. Ozarowski, L.-C. Brunel, H.-M. Stoeckli-Evans, J. Krzystek, Inorg. Chem. 2005, 44, 187.
| Crossref | GoogleScholarGoogle Scholar | 15651863PubMed |
[38] C. Duboc, D. Ganyushin, K. Sivalingam, M.-N. Collomb, F. Neese, J. Phys. Chem. A 2010, 114, 10750.
| Crossref | GoogleScholarGoogle Scholar | 20828179PubMed |
[39] M. Stebler, A. Ludi, H. B. Bürgi, Inorg. Chem. 1986, 25, 4743.
| Crossref | GoogleScholarGoogle Scholar |
[40] P. A. Goodson, J. Glerup, D. J. Hodgson, K. Michelsen, E. Pedersen, Inorg. Chem. 1990, 29, 503.
| Crossref | GoogleScholarGoogle Scholar |
[41] A. R. Oki, J. Glerup, D. J. Hodgson, Inorg. Chem. 1990, 29, 2435.
| Crossref | GoogleScholarGoogle Scholar |
[42] P. A. Goodson, J. Glerup, D. J. Hodgson, K. Michelsen, H. Weihe, Inorg. Chem. 1991, 30, 4909.
| Crossref | GoogleScholarGoogle Scholar |
[43] T. Yatabe, M. Kikkawa, T. Matsumoto, H. Nakai, K. Kaneko, S. Ogo, Dalton Trans. 2014, 43, 3063.
| Crossref | GoogleScholarGoogle Scholar | 24323354PubMed |
[44] P. A. Goodson, D. J. Hodgson, Inorg. Chem. 1989, 28, 3606.
| Crossref | GoogleScholarGoogle Scholar |
[45] K. J. Brewer, M. Calvin, R. S. Lumpkin, J. W. Otvos, L. O. Spreer, Inorg. Chem. 1989, 28, 4446.
| Crossref | GoogleScholarGoogle Scholar |
[46] G. R. Hanson, K. E. Gates, C. J. Noble, M. Griffin, A. Mitchell, S. Benson, J. Inorg. Biochem. 2004, 98, 903.
| Crossref | GoogleScholarGoogle Scholar | 15134936PubMed |
* Dedicated to Emeritus Professor Richard Robson (University of Melbourne), an outstandingly innovative and intelligent scientist, and a true gentleman, whose seminal research contributions to macrocyclic chemistry and metal organic frameworks are internationally renowned.