

Active Site Elucidation in Heterogeneous Catalysis via In Situ X-Ray Spectroscopies
Adam F. Lee Cardiff Catalysis Institute, School of Chemistry, Cardiff, UK.
Email: leeaf@cardiff.ac.uk
Australian Journal of Chemistry 65(6) 615-623 https://doi.org/10.1071/CH11455
Submitted: 2 December 2011 Accepted: 11 January 2012 Published: 9 February 2012
Abstract
Nanostructured heterogeneous catalysts will play a key role in the development of robust artificial photosynthetic systems for water photooxidation and CO2 photoreduction. Identifying the active site responsible for driving these chemical transformations remains a significant barrier to the design of tailored catalysts, optimized for high activity, selectivity, and lifetime. This highlight reveals how select recent breakthroughs in the application of in situ surface and bulk X-ray spectroscopies are helping to identify the active catalytic sites in a range of liquid and gas phase chemistry.
Introduction
Artificial photosynthesis offers an exciting approach to addressing two of this century's most difficult global challenges: the need for renewable energy sources to mitigate climate change and meet the demands of a growing world population; and provision of new routes to sustainable chemical feedstocks for e.g. plastics, polymers, and pharmaceuticals. To date, such needs have been met by (dwindling) fossil fuel reserves, hence there is now a powerful drive for alternative sources of fuels and chemicals. CO2 utilization as a feedstock for energy or chemicals synthesis is a particularly attractive strategy to ameliorate carbon emissions, while offering sustainable, safe, and useful carbon capture. Current CO2 utilization for chemical synthesis (principally urea) accounts for only 2 % of emitted CO2, but forecasts predict such approaches could mitigate 300–700 Mt (megatons) CO2 per year, far larger than the combined potential for CO2 abatement by nuclear, wind, and cellulosic biofuel technologies (~50 Mt CO2 per year).[1] Indeed the recent CS3 White Paper ‘A Sustainable Global Society’ highlights photocatalytic CO2 conversion to chemicals as an area where comprehensive fundamental materials chemistry research is essential.[2]
While there has been much recent attention focussed on so-called solar fuels production (principally methane or methanol), interest in direct routes to solar chemicals is growing. Olefins and their polymers are the single largest chemical commodity in the world, with global ethene and propene production capacity in 2010 estimated to be 123 and 77 Mt/year, respectively.[3] These olefins are obtained from non-renewable fossil fuels[4]: commercial ethene and propene manufacture involves steam or catalytic cracking of naphtha, gas oil and condensates to hydrocarbon mixtures followed by distillation. Cracking crude oil to produce ethene or propene is thermodynamically unfavourable (ΔG ≈ +100 kJ mol–1), requiring high temperatures (>600°C) to overcome the huge activation barriers to C-C cleavage (280 kJ mol–1 for kerosene conversion[5]). Hence steam cracking is the most energy-consuming process in chemistry, accounting for 8 % of the sector’s primary energy use and annual CO2 emissions of 180–200 Mt![4]
Harnessing solar energy to photoreduce CO2 to fuels or light olefins (e.g. 2CO2 + 2H2O ↔ C2H4 + 3O2), could create new chemical supply chains free of current dependencies on oil, coal, and natural gas (Fig. 1), but will require intensified processing and carefully engineered robust catalytic nanomaterials with high surface areas, able to activate small molecules at ambient conditions.[6] This challenge can only be met through novel nanotechnologies, which permit the construction of tailored materials whose structure (and function) can be precisely tuned.[7] Photocatalysis requires appropriately selected coupled redox reactions, and the ability to efficiently harness light capable of driving the requisite electron-transfer chemistry.[8] Heterogeneous (solid) catalysts will likely act as a critical component in future artificial photosynthetic systems,[9] due to their high physical and chemical tolerance towards e.g. temperature, pH, and irradiation compared with biological/bio-mimetic counterparts, and ability to absorb solar energy directly, or mediate it's transport from sensitizers[10]/light-harvesting antenna within hybrid bio-inorganic nanoarchitectures.
|
Despite a formal history dating back almost 200 years, since Humphrey Davy first reported catalytic combustion over Pt wire,[11] heterogeneous catalysis remains a field in which progress has largely resulted from empirical experimental observations in combination with serendipitous discoveries.[12] The absence of a unified theory of heterogeneous catalysis has largely reflected difficulties in (i) synthesizing well defined, high surface area materials possessing a narrow distribution of coordination environments and oxidation states, and (ii) identifying the active surface species participating in the catalytic cycle. While advances in top-down[13–16] and bottom-up[17–23] engineering of nanocrystals and nanoporous solids are helping to address the former, a limited ability to directly observe surface reactions, coupled with reaction-induced restructuring,[24] has hampered past efforts to fingerprint and quantify structure-function relationships even in simple gas phase heterogeneously catalyzed processes such as CO[25,26]/alcohol oxidation[27] and alkene reduction.[28] This highlight paper illustrates how recent technical breakthroughs in X-ray photoelectron spectroscopy (XPS), X-ray absorption spectroscopy (XAS) and X-ray emission spectroscopy (XES), are enabling researchers to visualize catalytic processes under in situ or operando (true working) conditions, and use the resulting insight to nanoengineer improved heterogeneous catalysts.
Surface Catalytic Ambient/High Pressure XPS
XPS has been a pivotal tool in the armoury of catalytic scientists for quantitatively probing the chemical environment (e.g. composition and oxidation state) of the outermost 1–10 atomic layers of catalyst surfaces under ultra-high vacuum (10–13 mbar). Over the past 15 years, time-resolved XPS[29] has helped unravel surface reaction mechanisms in precious metal catalyzed alcohol[30,31] and alkene[32,33] selective oxidation, C-C coupling,[34,35] dehalogenation,[36,37] and thermal[38–40] and photochemical[41] C-H activation, but has been restricted to studies of strongly-bound reactants over pristine model systems in the absence of solvents. Recent advances in surface science instrumentation, notably access to 3rd-generation synchrotron light sources and differentially-pumped, electron optics,[42] are now helping bridge the so-called ‘pressure gap’ in catalysis[12] and facilitate time-resolved XPS measurements at pressures up to 1 mbar,[43] sufficient to stabilize weakly bound adsorbates e.g. volatile liquids, and thereby simulate humid environments.[44] Such high pressure XPS (HP-XPS) or ambient pressure photoelectron spectroscopy (APPES) techniques also permit studies over thermodynamically unstable catalyst surface structures.[45] The power of APPES was recently demonstrated by Tao and co-workers to uncover new redox chemistry in bimetallic Pd@Rh core-shell nanoparticles.[46] Hitherto unexpected reversible palladium surface segregation was seen during coincident catalytic CO oxidation and NO reduction, key reactions in pollution abatement systems, with Rh migrating back to the surface under oxidizing NO environments (Fig. 2). In contrast, Pd (present predominantly as PdO) remains at the surface of Pt@Pd core-shell nanoparticles under oxidizing or reducing conditions. These in situ studies provide direct evidence that Pd/PdOx is the active component of such catalytic converters.
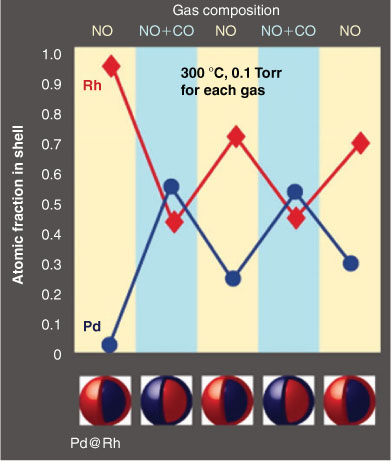
|
APPES has also been used to investigate the reactivity of NO and O2 over Au nanoparticles dispersed on silica or titania.[47] Au/TiO2 has aroused significant interest in photocatalysis as both a charge mediator for photo-excited electron transfer,[48,49] and as a light amplifier in e.g. CO2 photoreduction by water to ethene, methanol and formaldehyde,[50] wherein gold surface plasmons create intense local electromagnetic fields enhancing titania visible light photoactivity. Using APPES, Herranz demonstrated that only titania promoted a strong catalyst interaction with gas phase molecules; in situ measurements under 240 mTorr of NO revealed dissociative chemisorption resulting in NO3, dinitrosyl and atomic nitrogen reactive products exclusively on the TiO2 surface (Fig. 3), with the molecular adsorbates inducing band-bending.
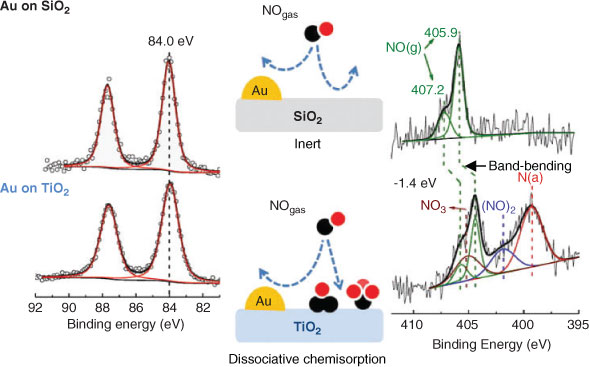
|
Product selectivity is an oft-neglected facet of catalytic transformations, wherein it can be particularly difficult to discriminate the desired selective catalytic centre from unselective counterparts.[51–53] In a recent HP-XPS study of alkyne hydrogenation over Pd,[28] Teschner demonstrated that the transition from selective to total hydrogenation of ethyne, propyne and 1-pentyne with increasing hydrogen pressures >1 mbar, coincided with the transformation from a Pd carbide phase, which inhibited dissolution of excessive hydrogen subsurface, to ‘bulk-like’ β-PdH (Fig. 4). Such structures can only be stabilized (and identified) via such in situ experiments, enabling unambiguous assignment of PdC as the requisite catalyst site for e.g. purification of ethene feedstreams for polymer production by selective removal of ethyne impurities. Further in situ XPS and XAS studies of propyne partial hydrogenation over ternary Cu-Ni-Fe catalysts,[54] show that Ni serves to enhance H2 activation, modulate Fe surface segregation and improve the synergy between iron and copper to deliver a catalyst formulation 100 % selective towards propene.
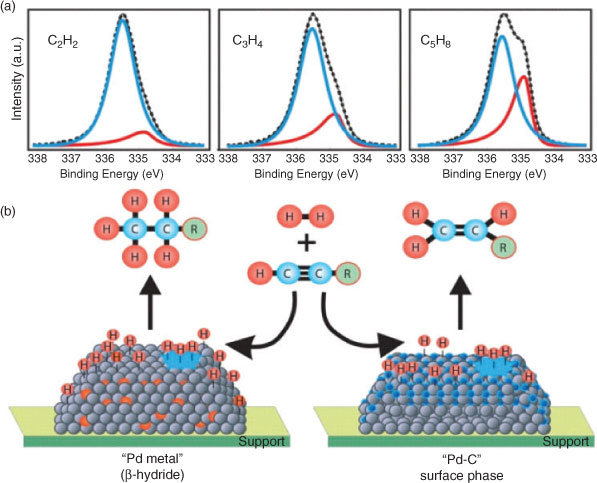
|
Operando Liquid and Gas Phase XAS
The timeline for in situ and operando XAS studies of catalytic processes (as opposed to e.g. thermal or reductive/oxidative pretreatments) parallels that of in situ XPS, with the first operando investigation of a heterogeneously catalyzed liquid phase reaction only reported in 2001.[55] XAS perfectly complements XPS in the study of catalytically active nanomaterials, probing the local chemical environment (and oxidation state) of the selected element of interest to derive an effective radial distribution function that maps the nature, number, and distance of surrounding atoms. However, since it is typically high-energy transmitted or fluorescent photons that are detected in XAS measurements, the technique can be utilized to monitor catalytic species within a variety of reaction cells containing windows of X-ray transmissive materials such as Si, BN or polyimide, and in weakly absorbing media including high pressures of gases or organic solvents. When operated in its conventional photons in-/photons-out mode, XAS is also a bulk-averaging method, although some surface sensitivity is possible in total electron yield mode.[56–58] There are many detailed articles and reviews on the application of XAS to heterogeneous catalysis[59–63] and photochemistry,[64–66] hence only illustrative examples on the breadth of systems/materials, and wealth of information on reaction mechanisms, available by time-resolved XAS approaches are provided.
The first detailed studies on liquid phase catalyst chemistry explored structure-function relations in the palladium catalyzed aerobic selective oxidation of crotyl and cinnamyl alcohols to their aldehydes,[27,51,67] important intermediates in fragrances, preservatives, and pesticides. Constructing simple, low-cost trickle-bed and recirculating reactors, our group used QuickXAS to demonstrate that catalyst deactivation was associated with in situ reduction of 2 nm oxidic Pd clusters to 10 nm metallic nanoparticles (Fig. 5), in stark contrast to the conventional wisdom of previous studies.[68] This insight directly led to the development of exceptionally active Pd catalysts comprising atomically-dispersed Pd2+ centres or stable PdOx nanoclusters on respective nanoporous alumina[69] and silica supports.[70] We subsequently extended this methodology to operando, time-resolved XAS studies of Pd nanoparticles during Suzuki-Miyaura cross-coupling, evidencing a surface-catalyzed contribution to the reaction mechanism.[35,71]

|
A significant problem likely to be encountered when constructing any photocatalytic reactor with macroscopic dimensions for practical artificial photosynthesis, is that of ensuring spatial homogeneity of integral nanostructured components, such as titania nanocrystals for CO2 photoreduction to methane, within a coating.[72–74] Scanning transmission X-ray microscopy (STXM) offers a powerful solution to this problem, and de Smit and co-workers recently implemented this technique employing soft X-rays (200–2000 eV) to image the active phase of an iron-based Fischer-Tropsch catalyst and reacting organic phase,[75] with a spatial resolution of 15 nm during the conversion of syngas (CO and H2) to hydrocarbons at 250°C and 1 bar pressure (Fig. 6). By comparing the spatial distribution of XAS spectra of reactively-formed hydrocarbons with those of different Fe-containing phases, they were able to demonstrate that iron carbide plays an important role in the catalytic cycle, with product spillover to the silica support preventing blocking of these active sites. Fluorescent μ-XAS has also been used to image an individual NiOx/Ce2Zr2Oy catalyst particle used in syngas production, enabling identification of the active Ni/Ce2Zr2O7 phase, and its spatial discrimination from inactive NiO/Ce2Zr2O8.[76]
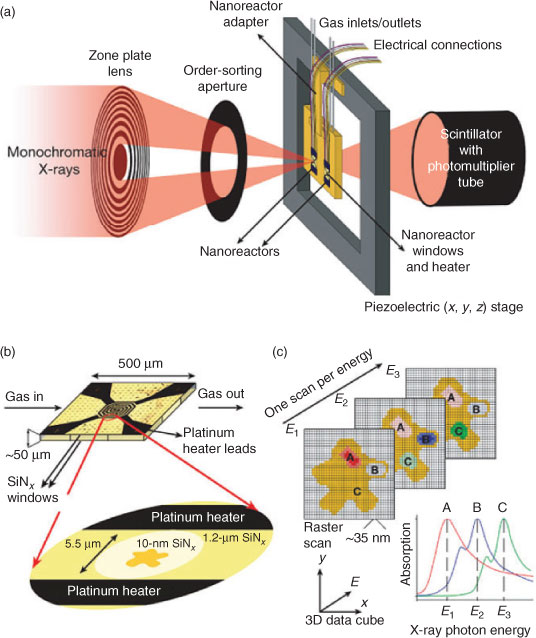
|
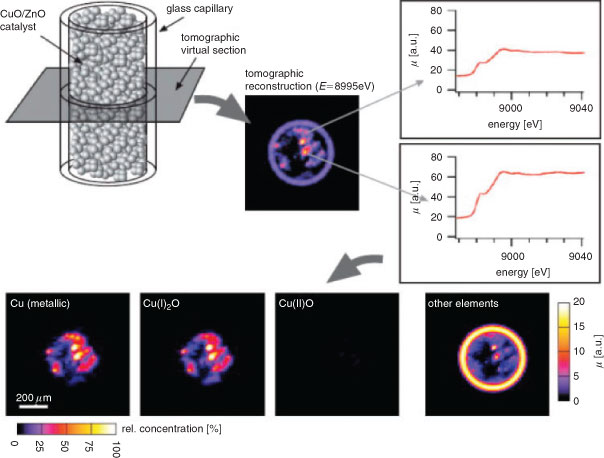
Scanning XAS microtomography offers a complementary approach,[77] in which instead of scanning a (micro)focussed X-ray beam across the catalyst, a position sensitive detector provides simultaneous full-field imaging across a section of the entire reactor bed as the incident X-ray energy is varied. This method provides the local chemical/electronic environment of selected elements with ~100 nm resolution. Subsequent sample rotation and tilting results in a complete 3D map of the distribution of different catalyst components or phases throughout a reactor bed (Fig. 7). 2D imaging has also been utilized to study the spatiotemporal evolution of reacting wavefronts of oxidized and reduced metal species within a Pt-Rh/Al2O3 catalyst, which respectively promote the total or partial oxidation of methane at 283°C.[78,79]
|
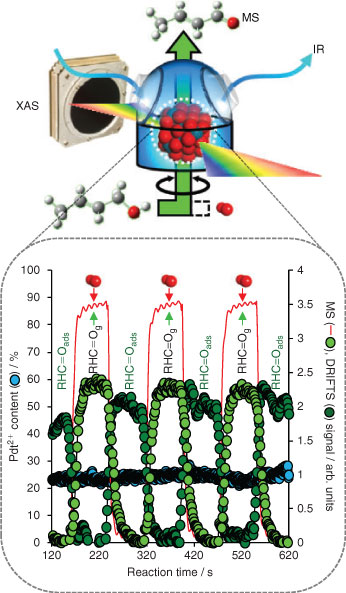
In situ XAS can be readily coupled with other analytical techniques,[80–83] such as diffraction, diffuse-reflectance infrared spectroscopy (DRIFTS), Raman,[84] or mass spectrometry (MS), to provide additional real-time insight into heterogeneous catalysts. The powerful combination of three spectroscopic techniques to probe the active site in heterogeneous catalysis under operando conditions was first demonstrated via energy-dispersive XAS/UV-Vis/Raman measurements of silica- and alumina-supported molybdenum oxide catalysts for propane dehydrogenation, enabling discrimination of two deactivation pathways via coking or MoO3 clustering, and tentative assignemt of Mo4+ as the active site.[85] However, in addition to ensuring excellent spatial homogeneity in any form of photocatalyst coating or packed bed, it is critical to ensure the catalyst thickness matches the penetration depth of the photoexciting light. Although there are several ways to facilitate light penetration throughout reactor beds, e.g. through the use of macroporous light-scattering voids, probing structure-function relations via coupled, multi-dimensional X-ray spectroscopies raises issues regarding each technique sampling the same catalyst volume.[83] We recently used synchronous energy-dispersive XAS/DRIFTS/MS to track the dynamic response of nanoparticulate Pd catalysts to changing reactive environments during the vapour phase aerobic selective oxidation of crotyl alcohol between 80 and 250°C.[53] Using an innovative reactor design,[86,87] catalyst oxidation state (XAS), reactively-formed surface intermediates (DRIFTS), and evolved gas phase products can be simultaneously followed on a sub-second timescale (Fig. 8). Adopting this transient approach uncovered a flexible (temperature sensitive) surface restructuring from Pd oxide to metal on contacting with crotyl alcohol, which could be reversed under dioxygen. We were also able to unequivocally differentiate the catalytic roles of PdO and metallic Pd: Pd2+ active sites drive the desired oxidative dehydrogenation pathway to crotonaldehyde; while Pd0 centres are responsible for aldehyde decarbonylation. Such structure-function relations could not have been delineated by conventional, steady-state XAS measurements. Time-resolved, in situ XAS has also been utilized to track (de)alloying in bimetallic Rh-Pd nanoparticles during gas phase NO reduction by H2, in which Pd surface enrichment helps suppress undesired N2O production via Rh oxidation.[88]
|
In Situ and Operando XES Studies
The near-edge structure of X-ray absorption spectra (XANES) can provide a direct map of the local empty density of states within condensed matter, such as metal or semiconductor nanoparticles employed in photocatalysis. However, intrinsic broadening renders experimental XAS spectra much broader than the actual density of states (DOS). Monitoring fluorescent photons, emitted after filling the X-ray excited core hole with valence electrons, offers improved spectral resolution, and the ability to discriminate the local environment of different valence states of an element within the same material and the interaction of active sites with adsorbates. There are several different modes of XES, which have been extensively reviewed elsewhere,[62] such as resonant inelastic X-ray scattering (RIXS), a powerful tool for studying the electronic structure of transition metal complexes in situ. RIXS was recently applied to elucidate the mechanism of CO poisoning of Pt nanoparticles[89] (an important process in CO2 photoreduction to hydrocarbons over Pt/TiO2 catalysts[90]), arising from the metal d-band shifting to lower energy. High-energy resolution fluorescence detected (HERFD) XAS utilizes the element-specific detection of selected fluorescence decay channels with long core-hole lifetimes to provide more detailed electronic and geometric structural information on catalytically active centres.[91] HERFD has been used to track the evolution of copper species within a working Cu/ZnO/Al2O3 methanol synthesis catalyst (Fig. 9) enabling Cu+ to be identified as an inert intermediate to the active and stable Cu0 species formed during in situ reduction.[92]

|
As with conventional XAS, XES can also be coupled with other spectroscopic and kinetic techniques for in situ catalyst characterization in both the liquid and gas phase. Van Bokhoven and co-workers recently combined HERFD-XAS with attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy to explore the solvent-dependent reduction of a HAuCl4 precursor deposited on ceria,[82] and subsequent correlation between gold oxidation state and reactive species present in solution during nitrobenzene hydrogenation. ATR-FTIR demonstrated that hydrogenation followed a stepwise mechanism, via formation of an azoxybenzene intermediate, with HERFD identifying only metallic gold (and not cationic) as the catalytically active species present throughout reaction (Fig. 10). It should prove possible to adopt this approach to investigate metal-semiconductor nanoparticle driven water photolysis.

|
Conclusion
Recent breakthroughs in optic, detector, and reactor cell designs, in conjunction with global access to high energy synchrotron radiation facilities, are transforming in situ X-ray spectroscopy into an exciting toolbox with which to probe the active site of working solid catalysts, operating over wide temperature and pressure regimes. Improvements in spatial (10–100 nm) and time (1 ns–1 s) resolution, and the use of multi-dimensional approaches have led to exciting discoveries of new reaction pathways, electron transfer processes, and hitherto unexpected catalytic phases. Such techniques are readily amenable to photocatalysis,[65,66] and will likely play an important role in developing (bio)inorganic nanostructured architectures for future solar fuels and chemicals synthesis.
Acknowledgements
AFL thanks the EPSRC for the award of a Leadership Fellowship (EP/G007594/2).
References
[1] M. Aresta, A. Dibenedetto, Dalton Trans. 2007, 2975.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXnsV2isLw%3D&md5=f3fa7eaedf5d45418b12e3fa3a2fd3f7CAS |
[2] http://www.rsc.org/images/sustainable-global-society-full-report_tcm18-200081.pdf.
[3] http://www.sriconsulting.com/WP/Public/Reports/propylene/.
[4] T. Ren, M. Patel, K. Blok, Energy 2006, 31, 425.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtFOmsLjJ&md5=30f8c9af94aa1ba23519236171cde313CAS |
[5] Y. Xing, W. Xie, W. Fang, Y. Guo, R. Lin, Energy Fuels 2009, 23, 4021.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXosVWmt7s%3D&md5=d932763f9bcf5747c5f25bebcf43c8b2CAS |
[6] S. C. Roy, O. K. Varghese, M. Paulose, C. A. Grimes, ACS Nano 2010, 4, 1259.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhs1Gmtrg%3D&md5=58c783ba69f3aa7b217cbc016e00c0f0CAS |
[7] V. L. Kuznetsov, P. P. Edwards, ChemSusChem 2010, 3, 44.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtVGnu7Y%3D&md5=4664ba158beb1651b94f093ef58f2500CAS |
[8] A. J. Morris, G. J. Meyer, E. Fujita, Acc. Chem. Res. 2009, 42, 1983.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVGntr3F&md5=53839fe8adaf6c01fa9c0c764850559fCAS |
[9] S. Linic, P. Christopher, D. B. Ingram, Nat. Mater. 2011, 10, 911.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFWqtb%2FO&md5=1ed74e57657948db6b0a8ce77ada7d5fCAS |
[10] C. Wang, R. L. Thompson, J. Baltrus, C. Matranga, The Journal of Physical Chemistry Letters 2009, 1, 48.
| Crossref | GoogleScholarGoogle Scholar |
[11] H. Davy, Phil. Trans. R. Soc. Lond. 1817, 107, 77.
| Crossref | GoogleScholarGoogle Scholar |
[12] G. Ertl, Angew. Chem. Int. Ed. 2008, 47, 3524.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXmt1Gitr0%3D&md5=b2ee9b3ce9b344499cd0c3997d8a1e69CAS |
[13] Y.-S. Hu, Y.-G. Guo, W. Sigle, S. Hore, P. Balaya, J. Maier, Nat. Mater. 2006, 5, 713.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XovFylurs%3D&md5=50145a29ba71727497e6d2e42cdb9771CAS |
[14] D. Y. Xia, D. Li, Z. Y. Ku, Y. Luo, S. R. J. Brueck, Langmuir 2007, 23, 5377.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXjvFyitrY%3D&md5=2e9d7692a06234f6cac697f84a9bfd48CAS |
[15] D. Verboekend, J. Perez-Ramirez, Catalysis Science & Technology 2011, 1, 879.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1SlsbbM&md5=f52dc3407c1ecab9858fd3b8a28be848CAS |
[16] T. Wakihara, A. Ihara, S. Inagaki, J. Tatami, K. Sato, K. Komeya, T. Meguro, Y. Kubota, A. Nakahira, Cryst. Growth Des. 2011, 11, 5153.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlSlurfF&md5=f27d9b784b3d3bfe7752313287dcad86CAS |
[17] Y. Wan, D. Zhao, Chem. Rev. 2007, 107, 2821.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXmslWgtbs%3D&md5=5328fe10008c3277e024ad1c28210071CAS |
[18] S. E. Habas, H. Lee, V. Radmilovic, G. A. Somorjai, P. Yang, Nat. Mater. 2007, 6, 692.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXpvFGqu7c%3D&md5=7445bd9adf5e42a95032266161e87c5eCAS |
[19] W. Fan, M. A. Snyder, S. Kumar, P. S. Lee, W. C. Yoo, A. V. McCormick, R. L. Penn, A. Stein, M. Tsapatsis, Nat. Mater. 2008, 7, 984.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsVWmu7nF&md5=e9ed3dbd5c3d7857adbd27a1eba46e3aCAS |
[20] J. P. Dacquin, J. Dhainaut, D. Duprez, S. Royer, A. F. Lee, K. Wilson, J. Am. Chem. Soc. 2009, 131, 12896.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVWnur3N&md5=8270067649690b45987d090bbe08f810CAS |
[21] V. Polshettiwar, R. S. Varma, Green Chem. 2010, 12, 743.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlvVGhsr4%3D&md5=b47f570aa9a3604238edf89541f8ed11CAS |
[22] J. Dhainaut, J. P. Dacquin, A. F. Lee, K. Wilson, Green Chem. 2010, 12, 296.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsleqsbs%3D&md5=b6caaf2b1114ceb4ba84928ac2cd9cb7CAS |
[23] C.-J. Jia, F. Schuth, Phys. Chem. Chem. Phys. 2011, 13, 2457.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVWjtr4%3D&md5=f7c39bf997a2ff9e93d950a59b1dd31dCAS |
[24] F. Tao, M. Salmeron, Science 2011, 331, 171.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjslSjuw%3D%3D&md5=eb83bbc91a69a44937748f9a7f9f19bbCAS |
[25] F. Cirak, J. E. Cisternas, A. M. Cuitino, G. Ertl, P. Holmes, I. G. Kevrekidis, M. Ortiz, H. H. Rotermund, M. Schunack, J. Wolff, Science 2003, 300, 1932.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXks1Kmu74%3D&md5=aad708e24272c691cbc668ce6a91246eCAS |
[26] M. A. Newton, C. Belver-Coldeira, A. Martinez-Arias, M. Fernandez-Garcia, Nat. Mater. 2007, 6, 528.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXnt1egsb4%3D&md5=f25079f85bccac55f6e975cb817bd614CAS |
[27] A. F. Lee, S. F. J. Hackett, J. S. J. Hargreaves, K. Wilson, Green Chem. 2006, 8, 549.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xltlars7s%3D&md5=5673e74af04a60d6dac54df8793ad99bCAS |
[28] D. Teschner, J. Borsodi, A. Wootsch, Z. Revay, M. Havecker, A. Knop-Gericke, S. D. Jackson, R. Schlogl, Science 2008, 320, 86.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXktVKhtrY%3D&md5=a76557b82a8d18b631b68d4ba65f5de2CAS |
[29] A. F. Lee, K. Wilson, R. L. Middleton, A. Baraldi, A. Goldoni, G. Paolucci, R. M. Lambert, J. Am. Chem. Soc. 1999, 121, 7969.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXltFSrsLc%3D&md5=0c6c62e4ba2e09c11cc93bc1271a6e17CAS |
[30] A. F. Lee, D. E. Gawthrope, N. J. Hart, K. Wilson, Surf. Sci. 2004, 548, 200.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXpslOrtbs%3D&md5=49eb3585ede471b2bb850ba2fab87f11CAS |
[31] A. F. Lee, Z. Chang, P. Ellis, S. F. J. Hackett, K. Wilson, J. Phys. Chem. C 2007, 111, 18844.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtlOntbnK&md5=de2b6041f75ce4e6fc8fe795a60c029dCAS |
[32] F. J. Williams, D. P. C. Bird, A. Palermo, A. K. Santra, R. M. Lambert, J. Am. Chem. Soc. 2004, 126, 8509.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXkvFyiurg%3D&md5=85b037f3756607273f9cea779cea7405CAS |
[33] R. L. Cropley, F. J. Williams, A. J. Urquhart, O. P. H. Vaughan, M. S. Tikhov, R. M. Lambert, J. Am. Chem. Soc. 2005, 127, 6069.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXivV2ntrw%3D&md5=e6c7e9f255072fc1b6054cc914b941adCAS |
[34] V. K. Kanuru, G. Kyriakou, S. K. Beaumont, A. C. Papageorgiou, D. J. Watson, R. M. Lambert, J. Am. Chem. Soc. 2010, 132, 8081.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmtlCrtbc%3D&md5=d5688e6425c9c7fa9f70ff1fc334b4bdCAS |
[35] A. F. Lee, P. J. Ellis, I. J. S. Fairlamb, K. Wilson, Dalton Trans. 2010, 39, 10473.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlGmsrfK&md5=cc11262751c5a2e30a10984572dbdda1CAS |
[36] A. F. Lee, K. Wilson, J. Phys. Chem. B 2006, 110, 907.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XlvFWjtA%3D%3D&md5=ce9542d3e7b79891eee4e4ad69097f7fCAS |
[37] A. F. Lee, Z. P. Chang, S. F. J. Hackett, A. D. Newman, K. Wilson, J. Phys. Chem. C 2007, 111, 10455.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXmslWgu7k%3D&md5=fa70448204cc82f4f8d4e338bc824d80CAS |
[38] A. F. Lee, K. Wilson, J. Vac. Sci. Technol. A 2003, 21, 563.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXjsVCrsrs%3D&md5=47c3a723d4518230dbb6fe6f293614e5CAS |
[39] T. Fuhrmann, M. Kinne, B. Tränkenschuh, C. Papp, J. F. Zhu, R. Denecke, H. P. Steinrück, New J. Phys. 2005, 7, 107.
| Crossref | GoogleScholarGoogle Scholar |
[40] C. Papp, B Tränkenschuh, R Streber, T Fuhrmann, R Denecke, H. P. Steinrück, J. Phys. Chem. C 2007, 111, 2177.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXltlaltA%3D%3D&md5=0e54516daba4451c53f6f61fdbaf9641CAS |
[41] R. Larciprete, A. Goldoni, A Groŝo, S Lizzit, G Paolucci, Surf. Sci. 2001, 482–485, 134.
| Crossref | GoogleScholarGoogle Scholar |
[42] D. F. Ogletree, H. Bluhm, G. Lebedev, C. S. Fadley, Z. Hussain, M. Salmeron, Rev. Sci. Instrum. 2002, 73, 3872.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xot1Whs7g%3D&md5=8f7a96da5da0f9b9d4242478a208fa69CAS |
[43] M. Salmeron, R Schlögl, Surf. Sci. Rep. 2008, 63, 169.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXksFKmtrk%3D&md5=016e9de6159585be309dfb81c847c95aCAS |
[44] H. Bluhm, J. Electron Spectrosc. Relat. Phenom. 2010, 177, 71.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXksFSgu7c%3D&md5=4ed6a5d7af0ee6a12c4814767dbb85a7CAS |
[45] A. F. Lee, V. Prabhakaran, K. Wilson, Chem. Commun. 2010, 46, 3827.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmsFSkurk%3D&md5=9c7a27377cd57282b0472a3b0177b9b4CAS |
[46] F. Tao, M. E. Grass, Y. Zhang, D. R. Butcher, J. R. Renzas, Z. Liu, J. Y. Chung, B. S. Mun, M. Salmeron, G. A. Somorjai, Science 2008, 322, 932.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlaltL%2FK&md5=7613620fee6f2e30f1ac2f1493ce91d9CAS |
[47] T. Herranz, X. Y. Deng, A. Cabot, Z. Liu, M. Salmeron, J. Catal. 2011, 283, 119.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht12jsLfF&md5=7adb0d75bf0c913c0bdf806e0560dd7fCAS |
[48] H. Tada, T. Mitsui, T. Kiyonaga, T. Akita, K. Tanaka, Nat. Mater. 2006, 5, 782.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtVWqsLvP&md5=7f81053824eeb906e7ca470a995bf64dCAS |
[49] V. Subramanian, E. E. Wolf, P. V. Kamat, J. Am. Chem. Soc. 2004, 126, 4943.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXisVCqsLY%3D&md5=c7ffb5a93116d998a0ee357f7489ca32CAS |
[50] W. B. Hou, W. H. Hung, P. Pavaskar, A. Goeppert, M. Aykol, S. B. Cronin, ACS Catal. 2011, 1, 929.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXosFyqsLY%3D&md5=f7c992e5061eaf9e43122f85186d27c4CAS |
[51] A. F. Lee, K. Wilson, Green Chem. 2004, 6, 37.
| Crossref | GoogleScholarGoogle Scholar |
[52] D. Ferri, C. Mondelli, F. Krumeich, A. Baiker, J. Phys. Chem. B 2006, 110, 22982.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtFCltLvK&md5=591e02f394cecbc3a3d9c80d68bc4b79CAS |
[53] A. F. Lee, C. V. Ellis, J. N. Naughton, M. A. Newton, C. M. A. Parlett, K. Wilson, J. Am. Chem. Soc. 2011, 133, 5724.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjvVSitLs%3D&md5=c38818f62613c5469386ff6210840517CAS |
[54] B. Bridier, J. Perez-Ramirez, A. Knop-Gericke, R. Schlogl, D. Teschner, Chem. Sci. 2011, 2, 1379.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnsVyhs7k%3D&md5=5e4e5b6d25521dd8d3bdba3681e56129CAS |
[55] A. F. Lee, in 221st National Meeting of the American Chemical Society, 2001, San Diego, COLL 187 (American Chemical Society: Washington, D.C.).
[56] G. D. Moggridge, T. Rayment, R. M. Ormerod, M. A. Morris, R. M. Lambert, Nature 1992, 358, 658.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XltlymtLs%3D&md5=252156b39de52a53043c6016124447bbCAS |
[57] S. L. M. Schroeder, G. D. Moggridge, T. Rayment, R. M. Lambert, J. Mol. Catal. Chem. 1997, 119, 357.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXislSjsrc%3D&md5=c5316f9b1ac49cf59cbacac3aa9a94cfCAS |
[58] M. Havecker, A. Knop-Gericke, H. Bluhm, E. Kleimenov, R. W. Mayer, M. Fait, R. Schlogl, Appl. Surf. Sci. 2004, 230, 272.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXkt1Cqtb0%3D&md5=925a7c92ab8083e55fff58a3ebabcef1CAS |
[59] M. A. Newton, A. J. Dent, J. Evans, Chem. Soc. Rev. 2002, 31, 83.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XhvFyjtrg%3D&md5=bb4727d00ba13db34587d3c27c07cd68CAS |
[60] J. Evans, A. Puig-Molina, M. Tromp, MRS Bull. 2007, 32, 1038.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXosVOhtQ%3D%3D&md5=c77cb2ac3c91d7854ac5d3a5aacbefa0CAS |
[61] S. R. Bare, T. Ressler, in Advances in Catalysis 2009, Volume 52, p. 339 (Eds B. C. Gates, H. Knozinger) (Elsevier Academic Press Inc.: San Diego, CA).
[62] J. Singh, C. Lamberti, J. A. van Bokhoven, Chem. Soc. Rev. 2010, 39, 4754.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVejsL%2FK&md5=bd0d31c3b82d1c9e24e8f92bc2dad069CAS |
[63] A. M. Beale, S. D. M. Jacques, B. M. Weckhuysen, Chem. Soc. Rev. 2010, 39, 4656.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVejsLzL&md5=ccdc27de9f2d2802fdfd5d46c0b3208aCAS |
[64] L. X. Chen, Annu. Rev. Phys. Chem. 2005, 56, 221.
| 1:CAS:528:DC%2BD2MXps1Grsbs%3D&md5=f4fe3760c82ec7b33c98cb8d601cb777CAS |
[65] X. Zhang, G. Smolentsev, J. Guo, K. Attenkofer, C. Kurtz, G. Jennings, J. V. Lockard, A. B. Stickrath, L. X. Chen, The Journal of Physical Chemistry Letters 2011, 2, 628.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXis1aku7Y%3D&md5=a5321c9a6f6d431db15382225bb9a66aCAS |
[66] L. X. Chen, W. J. H. Jäger, G Jennings, D. J. Gosztola, A. Munkholm, J. P. Hessler, Science 2001, 292, 262.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXivVyhtLs%3D&md5=3b7d38c738cc16d266a04cdc1a868a03CAS |
[67] J. D. Grunwaldt, M. Caravati, A. Baiker, J. Phys. Chem. B 2006, 110, 9916.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XktVSksb8%3D&md5=ff80ab542c7d0d2bafb1a0871c29ecf3CAS |
[68] T. Mallat, A. Baiker, Chem. Rev. 2004, 104, 3037.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXis1Cjtb8%3D&md5=df00272a72086a743578cc0b31e58e50CAS |
[69] S. E. J. Hackett, R. M. Brydson, M. H. Gass, I. Harvey, A. D. Newman, K. Wilson, A. F. Lee, Angew. Chem. Int. Ed. 2007, 46, 8593.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtl2rsrnF&md5=d7236088117eae95542c27753448a46eCAS |
[70] C. M. A. Parlett, D. W. Bruce, N. S. Hondow, A. F. Lee, K. Wilson, ACS Catal. 2011, 1, 636.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXlvVSlur0%3D&md5=c885b01ddcf4d1db32d2090212c00bd0CAS |
[71] P. J. Ellis, I. J. S. Fairlamb, S. F. J. Hackett, K. Wilson, A. F. Lee, Angew. Chem. Int. Ed. 2010, 49, 1820.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXivFantbc%3D&md5=74c5767505f1aab2bb46ea230590df51CAS |
[72] T. V. Nguyen, J. C. S. Wu, Sol. Energy Mater. Sol. Cells 2008, 92, 864.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXms1Wqtr8%3D&md5=4acc8b9c4843143063d1777913a29350CAS |
[73] A. Nishimura, N. Komatsu, G. Mitsui, M. Hirota, E. Hu, Catal. Today 2009, 148, 341.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVCqs7fN&md5=29a6a90924b94ef1f029c16fd381a5e7CAS |
[74] P. Y. Liou, S. C. Chen, J. C. S. Wu, D. Liu, S. Mackintosh, M. Maroto-Valer, R. Linforth, Energy & Environ. Sci. 2011, 4, 1487.
| 1:CAS:528:DC%2BC3MXms1Wiu7w%3D&md5=2a4e9c139cbb1570c952c3dbf93775ffCAS |
[75] E. de Smit, I. Swart, J. F. Creemer, G. H. Hoveling, M. K. Gilles, T. Tyliszczak, P. J. Kooyman, H. W. Zandbergen, C. Morin, B. M. Weckhuysen, F. M. F. de Groot, Nature 2008, 456, 222.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlKmtL7I&md5=e423463497ec07e8e0ba77075d07c6e1CAS |
[76] M. Tada, N. Ishiguro, T. Uruga, H. Tanida, Y. Terada, S.-i. Nagamatsu, Y. Iwasawa, S.-i. Ohkoshi, Phys. Chem. Chem. Phys. 2011, 13, 14910.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVSrtbnN&md5=7f5802badbaf84bf18f3364204a1deaaCAS |
[77] J.-D. Grunwaldt, B. Kimmerle, A. Baiker, P. Boye, C. G. Schroer, P. Glatzel, C. N. Borca, F. Beckmann, Catal. Today 2009, 145, 267.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXptV2muro%3D&md5=4862d75f7806cb57b48a4e5b6121cfa6CAS |
[78] J.-D. Grunwaldt, S. Hannemann, C. G. Schroer, A. Baiker, J. Phys. Chem. B 2006, 110, 8674.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xjt1Wlt7w%3D&md5=7e63bb6cf0b84e6da95b7596f5448d0aCAS |
[79] B. Kimmerle, J.-D. Grunwaldt, A. Baiker, P. Glatzel, P. Boye, S. Stephan, C. G. Schroer, J. Phys. Chem. C 2009, 113, 3037.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtFyrurk%3D&md5=ca05cd8245fe839ee3c82d72d0b5a621CAS |
[80] S. D. M. Jacques, O. Leynaud, D. Strusevich, P. Stukas, P. Barnes, G. Sankar, M. Sheehy, M. G. O’Brien, A. Iglesias-Juez, A. M. Beale, Catal. Today 2009, 145, 204.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXptV2mtbg%3D&md5=2cf55db8fab86c19dde75145f9e874b1CAS |
[81] U. Bentrup, Chem. Soc. Rev. 2010, 39, 4718.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVejs7fE&md5=5acffbfdd8e1209f534b81fdc2db1031CAS |
[82] M. Makosch, C. Kartusch, J. Sa, R. B. Duarte, J. A. van Bokhoven, K. Kvashnina, P. Glatzel, D. L. A. Fernandes, M. Nachtegaal, E. Kleymenov, J. Szlachetko, B. Neuhold, K. Hungerbuhler, Phys. Chem. Chem. Phys. 2012,
| Crossref | GoogleScholarGoogle Scholar |
[83] M. A. Newton, W. van Beek, Chem. Soc. Rev. 2010, 39, 4845.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVejs7fM&md5=1374f6b8e0f1fbb6eb5b5f3594865c13CAS |
[84] J.-D. Grunwaldt, N van Vegten, A Baiker, W van Beek, Journal of Physics: Conference Series 2009, 190, 012160.
[85] A. M. Beale, A. M. J. van der Eerden, K. Kervinen, M. A. Newton, B. M. Weckhuysen, Chem. Commun. 2005, 3015.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXlt12mtLY%3D&md5=813973c0d1ff285172dee29f8bf88195CAS |
[86] M. A. Newton, A. J. Dent, S. G. Fiddy, B. Jyoti, J. Evans, Catal. Today 2007, 126, 64.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXosVSjsLw%3D&md5=b9c2ba8329fb851867a5cca857e840c0CAS |
[87] M. Newton, Top. Catal. 2009, 52, 1410.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXoslGmsbY%3D&md5=60a31b62c0235d210ac93e1ee9bbb3a0CAS |
[88] M. A. Newton, B. Jyoti, A. J. Dent, S. Diaz-Moreno, S. G. Fiddy, J. Evans, ChemPhysChem 2004, 5, 1056.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXmtFGqu7k%3D&md5=f74023ddfcc44e983b0dd295248c1cddCAS |
[89] P. Glatzel, J. Singh, K. O. Kvashnina, J. A. van Bokhoven, J. Am. Chem. Soc. 2010, 132, 2555.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVGisLo%3D&md5=06acd0f54f0a72742f8adbfdbd4c9635CAS |
[90] O. K. Varghese, M. Paulose, T. J. LaTempa, C. A. Grimes, Nano Lett. 2009, 9, 731.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtV2ltLk%3D&md5=0a18fa04d8dc6298f63c0763498b02a8CAS |
[91] J. Singh, M. Nachtegaal, E. M. C. Alayon, J. Stötzel, J. A. van Bokhoven, ChemCatChem 2010, 2, 653.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXnsFyktr4%3D&md5=7b05bda17169a93070b9b5c30a40b017CAS |
[92] E. Kleymenov, J. Sa, J. Abu-Dahrieh, D. Rooney, J. A. van Bokhoven, E. Troussard, J. Szlachetko, O. V. Safonova, M. Nachtegaal, Catal. Sci. Technol. 2012, 2, 373.
| 1:CAS:528:DC%2BC38XosleltA%3D%3D&md5=0f1805f661e36e40113d1fbe9910a0f4CAS |