

Photoactive and Physical Properties of an Azobenzene-Containing Coordination Framework*
James S. Caddy A , Thomas B. Faust A , Ian M. Walton B , Jordan M. Cox B , Jason B. Benedict B , Marcello B. Solomon A , Peter D. Southon A , Cameron J. Kepert A and Deanna M. D’Alessandro A CA School of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia.
B Department of Chemistry, University at Buffalo, The State University of New York, Buffalo, NY 14260-3000, USA.
C Corresponding author. Email: deanna.dalessandro@sydney.edu.au
Australian Journal of Chemistry 70(11) 1171-1179 https://doi.org/10.1071/CH17215
Submitted: 20 April 2017 Accepted: 17 June 2017 Published: 8 August 2017
Abstract
A new three-dimensional coordination framework, [Zn4(tbazip)3(bpe)2(OH)2]·bpe·{solvent} (where bpe = 1,2-di(4-pyridyl)ethene) containing the novel photoactive ligand tbazip (tbazip = 5-((4-tert-butyl)phenylazo)isophthalic acid) has been synthesised and crystallographically characterised. The photoactivity of discrete tbazip was investigated and compared with its photoactivity while incorporated within the framework. The effect of isomerisation of the incorporated azobenzene on the chemical and physical properties of the framework were investigated using UV-vis and Raman spectroscopies. The framework is porous only to hydrogen gas at 77 K, but displayed an appreciable uptake for CO2 at 195 K.
Introduction
Functional inorganic materials that may be altered by the application of an external stimulus, such as pressure,[1] temperature,[2] or light,[3] have been rapidly developing over the past decade. Light stimulus is of particular interest because it is highly accessible, tunable, and may be used as a way to modify the structure of a functional inorganic material to induce a response (referred to as photoresponsivity).[4] A particular class of materials that has been at the centre of these studies is metal-organic frameworks (MOFs). The prospect of incorporating photoactivity into MOF systems is garnering significant attention at present, owing to the prospect of exploiting the light activity to perform useful functions such as ‘cargo delivery’,[5] tunable molecular separations,[6] controlled CO2 adsorption/desorption,[7–9] and catalysis (both heterogeneous[10] and photocatalysis[11]). A particular interest in controlled CO2 adsorption/desorption stems from the need to mitigate the excess CO2 released into the atmosphere, which has been identified as a global challenge.[12–16] This makes it particularly relevant to explore the interaction of different gases with a framework material.
One strategy to integrate photoactivity into a MOF is to exploit its tunability and to incorporate an organic linker that can change conformation upon light irradiation.[7,8,17,18] Photochromes are substances that undergo a chemical change upon exposure to electromagnetic radiation, forming products with significantly different absorption or emission spectra.[19] There exist many examples of photochromes in the literature, such as diarylethenes, spiropyrans, and azobenzenes.[20] MOFs incorporating both diarylethenes[18,21–23] and spiropyrans[24] have previously been reported. Azobenzenes in particular have gained significant attention and are very well studied because of their ability to undergo facile and rapid trans-cis isomerisation in high quantum yields around their central azo moiety upon exposure to UV light at 365 nm.[25] There lies great scope to functionalise the azobenzene core to immobilise it within a MOF; once incorporated, the change in geometry between the trans and cis isomers alters the distance of the two para carbons of the azobenzene from 9 to 5.5 Å, respectively,[26] which has great potential to affect the macroscopic properties of a MOF. The physical change is accompanied by an induced dipole moment change from 0 to 3 Da, respectively,[27] and a change in melting point of azobenzene.[28] An important property of these molecules is the reversibility of the light-induced changes, enabling the material to be used over multiple cycles.[29–31] The reverse reactions (from the cis to the trans form) require visible light irradiation or heating the sample.[32,33]
The reversible isomerisation properties of the azo functional group have received significant attention for inducing photoactivity in MOFs. Yanai et al. incorporated discrete azobenzene molecules into [Zn2(terephthalate)2(triethylenediamine)], where cis-trans isomerism altered the unit cell of the MOF from tetragonal to orthorhombic.[34] Hermann et al. performed similar studies on MOF-5, MIL-68(M) (M = Ga, In), and MIL-53(Al), where it was demonstrated that photoactive guests can only induce structural changes in MOFs if the pore size of the MOF allows for it, and if there is no dense packing of the azobenzene.[35] More recently, efforts have focussed on using visible light for the capture and release of CO2. Hill et al. have incorporated methyl red (an azobenzene derivative) into the pores of Mg-MOF-74 and MIL-53(Al), which assisted in the adsorption of CO2 after exposure to visible light.[9]
A strategy to control the density of azobenzene sites in MOFs has involved their installation as pendant groups onto ligands that form the backbone of the framework scaffold. This approach was first employed by Stock et al., who post-synthetically installed an azo functionality within a 4,4′-bipyridine (bpy) scaffold.[36] The same group subsequently reported the incorporation of an azo-based ligand into MIL-101-NH2(Cr),[37] whereby reversible light-induced cis-trans isomerisation of the azo bond occurred, although the reversibility was relatively low. A family of frameworks incorporating the azo-appended bpy scaffold have since been synthesised.[38] Subsequent work has investigated the effects of the switching of the azo group. In particular, Zhou et al. synthesised a MOF-5-type framework with appended azobenzenes which protruded into the pores.[39] Light dependent gas adsorption studies were undertaken, showing that the amount adsorbed was reliant on whether the cis or trans isomer was dominant in the material. The authors suggested that this difference was due to the pendant azo group blocking and unblocking access to the metal centre, respectively. MOFs incorporating azobenzenes have also been found to facilitate the trapping and release of dye molecules from the pores through a gating effect, where the movement of the azobenzene increases the mobility of the dye.[17] A similar paddling motion has been observed for the mechanism of CO2 uptake and release in azo-decorated isoreticular MOFs, where light induced isomerisation affects the adsorption of CO2.[7]
A further strategy for the development of photoresponsive MOFs is the direct incorporation of azobenzenes as pillaring ligands. In this case, the structural rigidity of a MOF may preclude the photoinduced change, or may initiate the degradation of the framework itself.[40] The direct incorporation of 4,4′-azobenzenedicarboxylic acid (abdc) into a UiO-67-type framework was reported by Schaate et al., although no cis-trans isomerisation could be observed in the MOF.[41] Lyndon et al.[8] investigated the framework [Zn(abdc)(bpe)], previously reported by Chen et al.[42] Here, a photoresponse was observed in the framework for the first time, where the azobenzene underwent cis-trans isomerisation in the pillars of the MOF, which led to measurable variations in the uptake and release of CO2. More recently, an azo-based MOF, Co3(dpia)2(azdc)3 {dpia = N1,N3-dipyridin-4-ylisophthalamide, azdc = 4,4′-diazene-1,2-diyldibenzoate} was reported by Gong et al.[43] who observed a breathing of the MOF upon irradiation with UV light, and CO2 capture and release performance of 45 % and 75 % under static and dynamic conditions, respectively. In addition to the findings of photoresponsive function, research has suggested that MOFs incorporating azobenzene ligands display an increased affinity for CO2,[44] and that azobenzene exhibits a degree of N2 phobicity that enhances its selectivity for CO2 over N2.[45]
Herein, we detail the synthesis and characterisation of a new coordination framework incorporating the novel azo-containing ligand 5-((4-tert-butyl)phenylazo)isophthalic acid. The photoactivity of both the ligand and the framework were investigated to gain insight into the modulation of the properties upon light activation. This work demonstrates some of the interesting effects of azo-incorporation into frameworks, and aids in the future development of novel multifunctional materials featuring functionalised azobenzene ligands. In the latter case, the ability to target porous structures with an optimal pore geometry is advantageous.
Experimental
Materials
All chemicals were obtained from Aldrich, Merck, Alfa Aesar, and Cambridge Isotope Laboratories (for deuterated solvents) and were used without further purification unless otherwise stated. 1H and 13C{1H} NMR spectra were recorded at 298 K on a Bruker AVANCE300, operating at 300 MHz for 1H and 75 MHz for 13C{1H}. Chemical shifts were standardised against published deuterated solvent shifts.[46]
Synthesis
5-(4-(tert-Butyl)phenylazo)isophthalic Acid (tbazip)
The method was adapted from that of Priewisch and Rück-Braun.[47] 1,3-Dimethyl-5-aminoisophthalate (2.1 g, 10 mmol) was dissolved in dichloromethane (80 mL). To this solution was added Oxone (12 g, 20 mmol) dissolved in water (250 mL). The two-phase mixture was stirred vigorously for 6 h under N2. The organic layer was removed and the aqueous layer was washed with dichloromethane (3 × 50 mL). The combined organic layers were washed with HCl (1 M, 200 mL), saturated aqueous NaHCO3 solution (200 mL), H2O (200 mL) and brine (200 mL), and dried over MgSO4. The solvent was removed by rotary evaporation, giving an impure yellow product. The unpurified 1,3-dimethyl-5-nitrosoisophthalate (1.5 g) was dissolved in glacial acetic acid with 4-tert-butylaniline (1.0 mL, 0.94 g, 6.3 mmol). The reaction was stirred at room temperature for 24 h. Ethyl acetate (EtOAc) (250 mL) was added, and the organic phase washed with copious water and sat. NaHCO3 solution then dried over MgSO4. The solvent was removed by rotary evaporation, giving an orange solid that was purified by silica column chromatography (cyclohexane 90 : 10 EtOAc) and washed with MeOH. The product, 5-(4-(tert-butyl)phenylazo)isophthalate (0.84 g, 2.4 mmol), was dissolved in a mixture of THF (70 mL), MeOH (10 mL), and aqueous NaOH solution (20 %, 10 mL). The reaction was refluxed at 50°C overnight, the organic solvent removed by rotary evaporation and the aqueous phase neutralised with HCl (1 M). The orange product was collected by vacuum filtration and dried. Yield: 0.79 g, 27 %. 1H NMR (300 MHz, [D6]DMSO) δ = 0.59 (s, 9H), 6.82 (d, 3JH-H = 9 Hz, 2H), 7.12 (d, 3JH-H = 9 Hz, 2H), 7.87 (d, 4JH-H = 1.5 Hz, 1H), 7.94 (t, 4JH-H = 1.5 Hz, 1H); 13C{1H} NMR (75 MHz, [D6]DMSO) δ = 22.1, 26.5, 114.6, 117.9, 118.9, 123.8, 124.5, 142.2, 144.8, 147,6, 158.8. FTIR ν(C=O) 1714, 1698 cm−1, ν(φ-N) 1281 cm−1, ν(N=N) 1448, 1411 cm−1. ESI-MS: (ESI+, MeOH) m/z calculated for C18H18N2O4 [M+H]+: 327.13, found: 327.09. Anal. Calc. for C18H18N2O4: C 66.25, H 5.56, N 8.58 %. Found: C 66.42, H 5.43, N 8.33 %.
[Zn4(tbazip)3(bpe)2(OH)2]·bpe·2H2O
5-(4-(tert-Butyl)phenylazo)isophthalic acid (33 mg, 0.10 mmol), 1,2-di(4-pyridyl)ethene (18 mg, 0.10 mmol), Zn(AcO)2·2H2O (22 mg, 0.10 mmol), KOH (5.5 mg, 0.10 mmol), EtOH (6 mL), and water (6 mL) were combined and sealed in a Teflon insert inside a Parr pressure reaction vessel. The vessel was heated at 120°C for 24 h and then cooled to 25°C over 24 h, giving a mixture of orange prismatic crystals and yellow powder, which was separated by density separation (CHCl3 7 : 1 EtOH) to give the crystalline product. Yield: 23 mg, 38 % based on tbazip. Anal. Calc. for [Zn4(tbazip)3(bpe)2(OH)2]·bpe·2H2O: C 58.39, H 4.57, N 9.08 %. Found: C 58.17, H 4.19, N 10.07 %.
Single Crystal X-Ray Diffraction (SCXRD)
X-Ray diffraction data for [Zn4(tbazip)3(bpe)2(OH)2]·bpe·{solvent} were collected using a Bruker SMART APEX2 CCD diffractometer installed at a rotating anode source (MoKα radiation, λ = 0.71073 Å), and equipped with an Oxford Cryosystems (Cryostream 700) nitrogen gas-flow apparatus. The data were collected at 90 K by the rotation method with 0.5° frame-width (ω scan) and 45 s exposure time per frame. Three sets of data (360 frames in each set) were collected, nominally covering the complete reciprocal space. Additional details regarding the data collection and refinement may be found in the Supplementary Material.
Crystal data for [Zn4(tbazip)3(bpe)2(OH)2]·bpe·{solvent}, C90H80N12O14Zn4 (M = 1815.14), CCDC 1538659: triclinic, space group P-1 (#2), a = 10.9673(7) Å, b = 16.0053(11) Å, c = 26.0812(18) Å, α = 91.5674(18)°, β = 94.1464(17)°, γ = 92.2275(17)°, V = 4560.6(5) Å3, Z = 2, T = 90.0 K, μ(MoKα) = 1.106 mm−1, Dcalc = 1.322 g/mm3, 75190 reflections measured (3.726 ≤ 2θ ≤ 69.974), 36791 unique (Rint = 0.0398, Rsigma = 0.0750) which were used in all refinements. The final R1 was 0.0560 (I >2σ(I)) and wR2 was 0.1485 (all data).
Powder X-Ray Diffraction (PXRD)
All data were collected on a PANalytical X’Pert PRO MPD diffractometer equipped with a PIXcel detector and CuKα radiation (λ = 1.5418 Å). Capillary PXRD measurements were conducted on samples under vacuum or a CO2 atmosphere. The samples were added to glass quartz capillaries and degassed overnight at either 373 or 403 K, then flame sealed with an oxygen micro-torch. Le Bail refinements were performed using GSAS.[48]
UV-vis-Near-Infrared Spectroscopy
All measurements were undertaken using a CARY 5000 UV-vis-near IR spectrophotometer. Solid state spectra were obtained using a diffuse reflectance Praying Mantis attachment and were recorded on a BaSO4 substrate against BaSO4 as a background standard. A Kubelka–Munk analysis was performed on the diffuse reflectance data. The KM factor, F(R) = (1–R)2/2R, where R is the diffuse reflectance of the sample as compared with the BaSO4 background was used to derive the absorption spectrum of the compound. Excitation was achieved in the dark using a 20 W Nelson Compact Fluorescent ultraviolet light bulb located 5 cm from the material. Solution state spectra were measured in EtOH. Excitation was achieved in the dark using a 4 W UV light Model ENF-240C/FE produced by Spectroline using the 365 nm wavelength setting located 5 cm from the sample.
Solid-State Fourier Transform Infrared Spectroscopy (FTIR)
FTIR spectra were collected using a Bruker Tensor 27 infrared spectrometer. Samples were measured in a dry KBr matrix against a KBr background for 64 scans over the range 400–4000 cm−1.
Resonance Raman Spectroscopy
Data were collected at the Mark Wainright Analytical Centre at the University of New South Wales using an inVia Raman Microscope equipped with an Argon ion laser exciting at 325 nm at × 40 resolution. All data were collected on single crystals and all measurements were composed of 10 static scans of 1 s duration.
Gas Adsorption
Adsorption isotherms over the 0–1 bar pressure range were recorded in glass analysis tubes using an ASAP2020 instrument from Micromeritics Instruments Inc. Each sample (40–90 mg) was degassed at either 373 or 403 K overnight before analysis. N2 isotherms were measured at 77 K using a liquid nitrogen bath and the Brunauer-Emmett-Teller (BET) surface area calculated. H2 isotherms were measured at 77 K in a liquid nitrogen bath. N2, CO2, O2, and H2 isotherms were also collected at 195 K using a dry ice and acetone bath. CO2 isotherms were also collected at 298 K using a circulating water bath.
Thermogravimetric Analysis (TGA)
Measurements were performed on a TA Instruments Hi-Res TGA 2950. Each sample (2–3 mg) was loaded onto a tared platinum sample pan and its mass recorded. The sample equilibrated for 10 min at 25–30°C and then heated to 600°C at 2°C min−1 under a constant N2 flow rate of ~0.1 L min−1.
Results and Discussion
Ligand Synthesis
The ligand tbazip was synthesised employing a similar method to that reported by Park et al.[27] Amino isophthalate was first oxidised to a nitroso compound using Oxone. The azo bond was formed by an acid catalysed Mills reaction between the isophthalate and tert-butyl aniline. The resultant esterified azo group was then deprotected to form the dicarboxylic acid. This procedure provides an effective pathway to generate a family of azo-functionalised isophthalic acids, as the same nitroso isophthalate may be reacted with a large number of substituted anilines.
Trans-cis Isomerisation of Discrete tbazip
Solid state UV-vis diffuse reflectance spectroscopy showed that tbazip exhibits the characteristic bands for a trans-azo compound (Fig. S1, Supplementary Material). Specifically, it features an intense band centred at 347 nm (28850 cm−1), assigned as a π-π* transition, and a relatively less intense band at 456 nm (21990 cm−1), which is assigned as the n-π* transition. Additional peaks are observed below 285 nm and are attributed to higher energy π-π* transitions. The spectrum for the ligand is similar to that reported for azobenzene itself, which features an n-π* transition at 457 nm (21900 cm−1) and a π-π* transition centred at 325 nm (30750 cm−1).[49]
UV light dependent studies were undertaken on tbazip to establish its photoactivity. Photoirradiation of tbazip with UV light led to a marked change in band intensities corresponding to trans-cis isomerisation. Upon excitation with a 365 nm light source, tbazip was partially excited from its ground trans to excited cis state, as shown by the increase in intensity of the n-π* band, and its blue shift from 441 to 435 nm (22660 to 22980 cm−1) (Fig. 1a). The reaction reached its photostationary state after 60 min. The decay of the excited state of tbazip was also monitored by UV-vis spectroscopy (Fig. 1b). The cis isomer was relatively stable in the dark, returning to its initial state after 120 min. When exposed to ambient light, however, cis-trans isomerisation occurred rapidly, with significant changes in the band intensities observed within 1 min and the isomerisation was complete within 20 min.

|
Raman studies were also undertaken to observe the isomerisation of tbazip in situ. As cis and trans azobenzenes possess different Raman spectra, it is possible to identify the ratio of isomers present.[50] A 325 nm laser was employed as this wavelength lies in the π-π* excitation band of tbazip. Spectra were recorded before and after irradiation of the ligand with UV light for 60 s (Fig. 2). Upon excitation, all peaks in the Raman spectrum decreased relative to the peak at 1605 cm−1, which also appeared to broaden significantly. Several studies of asymmetric azobenzenes have suggested that the decrease in relative intensity of peaks between 1400 and 1550 cm−1 is indicative of an isomerisation process.[51,52]
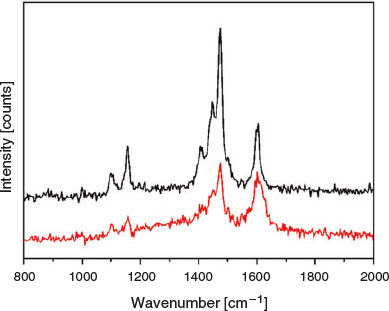
|
Crystal Structure and Thermophysical Properties of [Zn4(tbazip)3(bpe)2(OH)2]·bpe·{solvent}
The solvothermal reaction of tbazip, bpe, and zinc acetate under basic conditions yielded large orange crystals of [Zn4(tbazip)3(bpe)2(OH)2]·bpe·{solvent}. The asymmetric unit consists of four zinc centres, three deprotonated tbazip ligands, three bpe molecules, and two μ3-OH- ions (Fig. S3, Supplementary Material). Two of the zinc centres are six-coordinate and adopt octahedral configurations, while the remaining two zinc atoms are four-coordinate and exhibit a tetrahedral geometry. The coordination sphere of the tetrahedral zinc centres consists of a nitrogen atom from a pyridyl group and three oxygen atoms, two from two carboxylate groups and one from the hydroxide ion. The pyridyl nitrogen atom and a hydroxide ion are axially coordinated to the octahedral zinc centres while the equatorial sites consist of three oxygen atoms from three carboxylate groups and an additional hydroxide ion (Fig. S3, Supplementary Material).
The core of the zinc secondary building unit (SBU) is reminiscent of a typical paddlewheel SBU where the two metal centres are bridged equatorially by four carboxylate groups. In the present work, the metal centres form an ‘extended paddlewheel’ unit in which the ends of the SBU feature four carboxylate groups bridging two zinc centres. The ‘extension’ of the paddlewheel arises from the insertion of monodentate carboxylate groups that hydrogen bond to hydroxide ions located in the interior of the SBU (Fig. 3). The hydroxide ions in the centre of the cluster form an open parallelogram structure as they bridge between two octahedral and one of the tetrahedral zinc atoms. This M-OH bonding is similar to that described in several recent papers.[53,54]
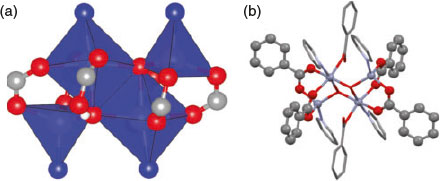
|
The low symmetry 3D framework exhibits three distinct bonding motifs along three different directions in the lattice (Fig. S4, Supplementary Material). Along the a- and c-axes, the metal SBUs are bridged by three crystallographically unique tbazip ligands coordinated to zinc atoms. Along the c-axis, both carboxylate groups of one of the unique tbazip ligand bridge two zinc atoms. The two remaining unique linkers exhibit identical connectivity along the a-axis. In both linkers, one of the carboxylate groups is bridging while the other exhibits monodentate binding. The oxygen atom of the monodentate carboxylate group that is not coordinated to a metal centre exhibits hydrogen bonding to the hydroxide group of the SBU (Fig. 4).
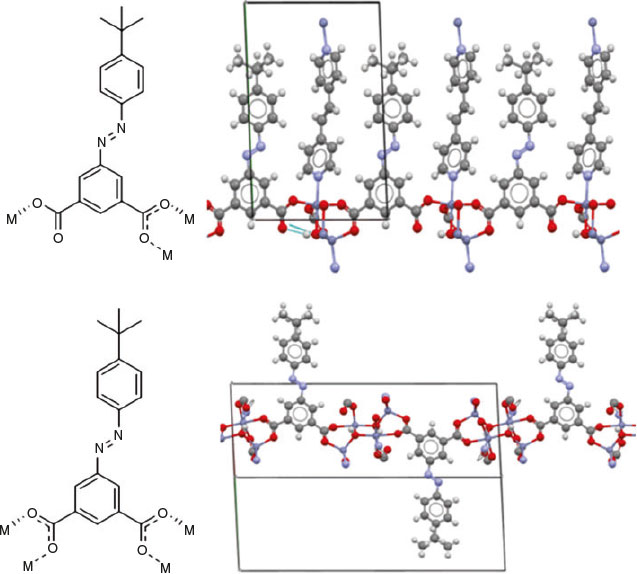
|
Connectivity along the b-axis occurs through the Zn-N bonds of a bpe molecule coordinated to two zinc atoms. This forms alternate layers of organic ligand and a mixed layer of the zinc cluster and bridging ligands (Fig. 5). The organic ligands feature extensive π-stacking interactions within the organic layers, and the free bpe molecule is held within the pores by π-stacking with nearby tbazip ligands. The crystal lattice contains regions occupied by disordered solvent molecules, the structures of which could not be reliably refined. The spaces occupied by these solvent molecules are connected to neighbouring solvent spaces to form channels along the a-axis. The observed porosity in [Zn4(tbazip)3(bpe)2(OH)2]·bpe·{solvent} is likely attributable to the partial retention of these channels upon evacuation of the solvent molecules.

|
PXRD analysis was employed to confirm the bulk purity of [Zn4(tbazip)3(bpe)2(OH)2]·bpe·2H2O (Fig. S5, Supplementary Material). Additionally, it was found that the PXRD pattern undergoes only relatively minor changes upon desolvation of the framework and upon subsequent exposure to 750 mbar CO2 (Fig. S9, Supplementary Material).
As the solvent could not be refined satisfactorily by SCXRD (the SQUEEZE[55] function of OLEX2[56] was used), thermogravimetric analysis (TGA) was used both to determine the amount of solvent in the sample and to estimate the porosity of the framework. TGA indicates that there is ~6.4 % w/w mass loss below 130°C; this is attributed to the loss of the disordered chemical species residing within the 594.8 Å3 of solvent accessible void space per unit cell (13.0 % v/v). The mass loss over the range 200–320°C is attributed to the loss of the bpe molecule occluded within the pores of the framework, which drives decomposition of the material. Supercritical CO2 drying was undertaken in an attempt to remove this ligand without damaging the framework; however, TGA analysis following the process still indicated the presence of excess ligand (Fig. S6, Supplementary Material).
Gas Adsorption
At 77 K, the desolvated form of the framework, [Zn4(tbazip)3(bpe)2(OH)2]·bpe, was found to be essentially non-porous to nitrogen gas (Fig. 6a), with a nominal BET surface area calculated at 30 m2 g−1. Hydrogen gas is able to diffuse into the structure, although the very slow adsorption/desorption kinetics cause substantial hysteresis. This behaviour is consistent with the narrow one-dimensional channels observed in the crystal structure (between 2.68 and 6.65 Å between adjacent hydrogen atoms on tbazip ligands).

|
At 195 K, the nitrogen gas uptake is greater (Fig. 6b), suggesting ‘gate-opening’ processes driven either by expansion of the channels or thermal excitation increasing the diffusion rate. Oxygen gas is adsorbed, and CO2 condenses in the micropore channels in a typical type I isotherm. The H2 uptake is relatively lower than that at 77 K due to the higher temperature.
The CO2 isotherm at 298 K (Fig. 6c) also demonstrates that the framework admits this guest, with a maximum uptake of 2.9 mmol g−1, or 12 % w/w at 1100 mbar. This uptake could be explained by the free bpe molecules within the pores, since accessible nitrogen atoms have been reported to bind CO2 effectively.[12] Additionally, simulations on the azo functional group suggest that interactions with CO2 are improved, compared with N2.[25]
Photoactivity of the Framework
The UV-vis diffuse reflectance spectrum of [Zn4(tbazip)3(bpe)2(OH)2]·bpe·2H2O strongly resembles that of the tbazip ligand. The main difference between the two spectra is the hypsochromic shift of the n-π* and π-π* bands by 10 nm (520 cm−1) and 18 nm (1360 cm−1), respectively (Fig. 7a).

|
Irradiation of the framework with UV light leads to an increase in the intensity of the n-π* band relative to the π-π* band, suggesting that trans-cis isomerisation is occurring (Fig. 7b). The change in the ratios begins to plateau after 120 min of irradiation, suggesting that a photostationary state was attained. Similarly, there are strong indications of the reverse cis-trans reaction occurring when light irradiation is ceased, through the observation of an isosbestic point at ~390 nm (25700 cm−1), in addition to the decrease in intensity of the n-π* band and increase of the π-π* band. There was a small change in intensity as the framework was rested in the dark for 110 min (Fig. 7c), but when exposed to ambient light, changes occurred more rapidly (Fig. 7d). It must be noted that the photochromic behaviour observed is likely to be confined to the surface of the particles.
Raman studies were also performed on the framework both before and upon irradiation with UV light at 325 nm for up to 120 s (Fig. 8). As was observed with the discrete tbazip, all peaks decreased relative to the peak at 1605 cm−1, which underwent broadening. This may suggest that surface isomerisation could be occurring; however, this peak change could also be a sign of crystal degradation.
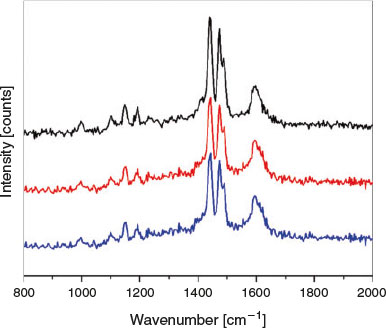
|
Note that in situ light dependent gas adsorption measurements were attempted; however, the changes in adsorption were not outside experimental uncertainty, and are therefore not reported here. Presumably, the densely packed nature of [Zn4(tbazip)3(bpe)2(OH)2]·bpe precludes the effect of isomerisation on the gas adsorption properties.
Conclusions
The new coordination framework [Zn4(tbazip)3(bpe)2(OH)2]·bpe·{solvent} containing the novel tbazip ligand was synthesised. UV-vis studies of both the ligand and its MOF following photoirradiation with UV light revealed an isomerisation process within the material. This process was found to be reversible in the solid state, with trans-cis isomerisation occurring after 15 min exposure to light. Upon the removal of light, the reverse cis-trans isomerisation was observed.
Gas adsorption studies at 77, 195, and 298 K revealed that the MOF exhibited a nominal BET surface area of 30 m2 g−1, but was found to adsorb CO2 despite its modest N2 uptake and surface area. It is postulated that the presence of the azo groups in the material aid in the uptake CO2, due to their positive calculated interaction with CO2 and their suggested ‘N2-phobicity’.[33] This work demonstrates some of the interesting effects of azo-incorporation into frameworks, and aids in the future development of novel multifunctional materials featuring functionalised azobenzene ligands.
Supplementary Material
Additional data for the crystallographic analysis, powder diffraction, and thermogravimetric analysis, as well as infrared and UV-vis-NIR data for the ligand and framework are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by the Australian Research Council and the University at Buffalo, the State University of New York. We gratefully acknowledge A/Prof. Matthew Hill and Dr Richelle Lyndon for their assistance in trialling the in situ light irradiated gas adsorption measurements on the framework, and Dr Samuel Duyker for his assistance with performing Le Bail refinements.
References
[1] K. J. Gagnon, C. M. Beavers, A. Clearfield, J. Am. Chem. Soc. 2013, 135, 1252.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXns1ejtA%3D%3D&md5=2a6d47487661536a730ffc1c01dd07dbCAS |
[2] S. Nagata, K. Kokado, K. Sada, Chem. Commun. 2015, 51, 8614.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXmsVais7w%3D&md5=59585ec7d9ac55b66f3a46d96c8f73a6CAS |
[3] J. Yu, Y. Cui, C.-D. Wu, Y. Yang, B. Chen, G. Qian, J. Am. Chem. Soc. 2015, 137, 4026.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXksl2gtrc%3D&md5=e4d4931069151b4d485703d30269178dCAS |
[4] F.-X. Coudert, Chem. Mater. 2015, 27, 1905.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXisF2hurw%3D&md5=56ffa9c423f8d7e68e6b82d1a87d07c3CAS |
[5] X. Meng, B. Gui, D. Yuan, M. Zeller, C. Wang, Sci. Adv. 2016, 8, e1600480.
| Crossref | GoogleScholarGoogle Scholar |
[6] Z. Wang, A. Knebel, S. Grosjean, D. Wagner, S. Bräse, C. Wöll, J. Caro, L. Heinke, Nat. Commun. 2016, 7, 13872.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XitFGjtbnM&md5=9d8bee4761ad69849a86f0bd25eb6d61CAS |
[7] R. Huang, M. R. Hill, R. Babarao, N. V. Medhekar, J. Phys. Chem. C 2016, 120, 16658.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XhtFeku77F&md5=77d284aea44636a5206d3ff4a8da6f42CAS |
[8] R. Lyndon, K. Konstas, B. P. Ladewig, P. D. Southon, P. C. J. Kepert, M. R. Hill, Angew. Chem. Int. Ed. 2013, 52, 3695.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXitlahur8%3D&md5=7e5654122375f9202b23e844e1d50c6fCAS |
[9] R. Lyndon, K. Konstas, A. W. Thornton, A. J. Seeber, B. P. Ladewig, M. R. Hill, Chem. Mater. 2015, 27, 7882.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXhslyns7%2FJ&md5=b8fdee356f7255fad721a5e38d087669CAS |
[10] L. T. M. Hoang, L. H. Ngo, H. L. Nguyen, H. T. H. Nguyen, C. K. Nguyen, B. T. Nguyen, Q. T. Ton, H. K. D. Nguyen, K. E. Cordova, T. Truong, Chem. Commun. 2015, 51, 17132.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXhsFKhtLjO&md5=84f171e84bf10449f7f2fba3d6869b13CAS |
[11] X. Sun, Q. Yu, F. Zhang, J. Wei, P. Yang, Catal. Sci. Technol. 2016, 6, 3840.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXitVOjsbfO&md5=07b16df21605717986a53090ad3e5c76CAS |
[12] D. M. D’Alessandro, B. Smit, J. R. Long, Angew. Chem. Int. Ed. 2010, 49, 6058.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtVaqu7fL&md5=7bff7ffdd10ba29fea6a418c29a5f77aCAS |
[13] A. Das, D. M. D’Alessandro, CrystEngComm 2015, 17, 706.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhsVSmtLrM&md5=0fb17966021463153780bc20f8378a5cCAS |
[14] E. S. Rubin, J. E. Davison, H. J. Herzog, Int. J. Greenh. Gas Control 2015, 40, 378.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXhtVegs7fM&md5=a98c09ab04e2c1348e9aa06d6793ebd0CAS |
[15] D. M. Thomas, J. Mechery, S. V. Paulose, Environ. Sci. Pollut. Res. Int. 2016, 23, 16926.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XhtF2hsrzK&md5=3d60488fd50a9b841bee28efdb666b64CAS |
[16] Z. Zhang, Z.-Z. Yao, S. Xiang, B. Chen, Energy Environ. Sci. 2014, 7, 2868.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhtVymu7rI&md5=6906e9d92dbc4654fad259023eb5f020CAS |
[17] J. W. Brown, B. L. Henderson, M. D. Kiesz, A. C. Whalley, W. Morris, S. Grunder, H. Deng, H. Furukawa, J. I. Zink, J. F. Stoddart, O. M. Yaghi, Chem. Sci. 2013, 4, 2858.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXos1yqtrY%3D&md5=7d2444a01201e378ce7195c83d8380b9CAS |
[18] C. B. Fan, Z. Q. Liu, L. L. Gong, A. M. Zheng, L. Zhang, C. S. Yan, H. Q. Wu, X. F. Feng, F. Luo, Chem. Commun. 2017, 53, 763.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XitFalsL7F&md5=4a163d3b6c133c1b411f80975feceeeeCAS |
[19] A. V. El’tsov, Organic Photochromes 1990 (Consultants Bureau: New York, NY).
[20] S.-L. Huang, T. S. A. Hor, G.-X. Jin, Coord. Chem. Rev. 2017, 346, 112.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XhtVSrs73I&md5=e79e391a23d55bf01c547352aae0ac7eCAS |
[21] J. M. Cox, I. M. Walton, J. B. Benedict, J. Mater. Chem. C 2016, 4, 4028.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28Xit1Oru7k%3D&md5=b585eb8c44e4f177e38a3de4fe668688CAS |
[22] I. M. Walton, J. M. Cox, C. A. Benson, D. G. Patel, Y.-S. Chen, J. B. Benedict, New J. Chem. 2016, 40, 101.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXhsFamtrzM&md5=dea6041f14089ecb96cb8b50c0165aebCAS |
[23] I. M. Walton, J. M. Cox, J. A. Coppin, C. M. Linderman, D. G. Patel, J. B. Benedict, Chem. Commun. 2013, 49, 8012.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXht1OjurfK&md5=86ad8517b31b2baa8b62d444ba20ac69CAS |
[24] K. Healey, W. Liang, P. D. Southon, T. L. Church, D. M. D’Alessandro, J. Mater. Chem. A 2016, 4, 10816.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XhtFSgtbbN&md5=d6ea555b235c34007a769ce32ebc2b38CAS |
[25] Y. Dou, Y. Hu, S. Yuan, W. Wu, H. Tang, Mol. Phys. 2009, 107, 181.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXkvVShurs%3D&md5=70b0165ec15dfb0aa4701b5110d42334CAS |
[26] H. Koshima, N. Ojima, H. Uchimoto, J. Am. Chem. Soc. 2009, 131, 6890.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXlsFCgu70%3D&md5=0c91f022d9283c8f717ee53f70a9f1f2CAS |
[27] G. S. Hartley, R. J. W. Le Fevre, J. Chem. Soc. (Res.) 1939, 531.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaA1MXjslKgsw%3D%3D&md5=5c8373640af5df8dcc845651cf8550b5CAS |
[28] S. Eligehausen, S. M. Sarge, G. Öhlschläger, H. K. Cammenga, J. Therm. Anal. 1989, 35, 515.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1MXmsVWhtL0%3D&md5=c5d243a8be98000da09cbc6ce479098dCAS |
[29] G. D. Jaycox, J. Polym. Sci. Part A: Polym. Chem. 2004, 42, 566.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXotFGlsg%3D%3D&md5=f1463a629ec0951ba47015ac69bbd7dcCAS |
[30] J. Shi, B. Wang, H. Du, Colloid Polym. Sci. 2014, 292, 1217.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXjtlSktLg%3D&md5=25e5b548590c696d86c1dbcf707ac211CAS |
[31] E. Vaselli, C. Fedele, S. Cavalli, P. A. Netti, ChemPlusChem 2015, 80, 1547.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXhtFWgu7fL&md5=88f275e28dc115f7181ff8af884149a0CAS |
[32] X.-P. Qiu, E. V. Korchagina, J. Rolland, F. M. Winnik, Polym. Chem. 2014, 5, 3656.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXns1WisbY%3D&md5=673e016596352d8bbf6e633728dffb97CAS |
[33] H. M. D. Bandara, S. C. Burdette, Chem. Soc. Rev. 2012, 41, 1809.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XitFyjsLg%3D&md5=3d372d80025ffe9eea833f1085407d6dCAS |
[34] N. Yanai, T. Uemura, M. Inoue, R. Matsuda, T. Fukushima, M. Tsujimoto, S. Isoda, S. Kitagawa, J. Am. Chem. Soc. 2012, 134, 4501.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XjtVCrur0%3D&md5=4d5b5b52dac2fea22dcd3bc727cd0490CAS |
[35] D. Hermann, H. Emerich, R. Lepski, D. Schaniel, U. Ruschewitz, Inorg. Chem. 2013, 52, 2744.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXisV2isbo%3D&md5=3f9f295ffd2c22cd20151fbcf3cd974fCAS |
[36] A. Modrow, D. Zargarani, R. Herges, N. Stock, Dalton Trans. 2011, 40, 4217.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXkt1Kgtrg%3D&md5=d91aa1cfb54242e0cf2f932849536a26CAS |
[37] A. Modrow, D. Zargarani, R. Herges, N. Stock, Dalton Trans. 2012, 41, 8690.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xpt1Cmu7k%3D&md5=635b6015751fe23ff854b5a394c99c30CAS |
[38] A. Modrow, M. Feyand, D. Zargarani, R. Herges, N. Stock, Z. Anorg. Allg. Chem. 2012, 638, 2138.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XnsleisrY%3D&md5=0ccae7c859ddbef5feacc147ee64b3deCAS |
[39] J. Park, D. Yuan, K. T. Pham, J. R. Li, A. Yakovenko, H. C. Zhou, J. Am. Chem. Soc. 2012, 134, 99.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1SmtbnP&md5=93ade79cd26277a5a4c6ff7c77f4e49cCAS |
[40] C. L. Jones, A. J. Tansell, T. L. Easun, J. Mater. Chem. A 2016, 4, 6714.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXitVCqsrvO&md5=31c31b235d9b604a7c56a4089ce5e032CAS |
[41] A. Schaate, S. Dühnen, G. Platz, S. Lilienthal, A. M. Schneider, P. Behrens, Eur. J. Inorg. Chem. 2012, 790.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XjvVGntQ%3D%3D&md5=cd268ebce9e7fe7f87fa091761b893f7CAS |
[42] B. Chen, S. Ma, E. J. Hurtado, E. B. Lobkovsky, H.-C. Zhou, Inorg. Chem. 2007, 46, 8490.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtVegsr%2FF&md5=ee62d8a92a7df4538107fbb1c3a8b204CAS |
[43] L. L. Gong, X. F. Feng, F. Luo, Inorg. Chem. 2015, 54, 11587.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXitVSnsL7N&md5=df090d31e15f63eefcf82159d6f0779cCAS |
[44] C. M. Nagaraja, R. Haldar, T. K. Maji, C. N. R. Rao, Cryst. Growth Des. 2012, 12, 975.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xpt1Wi&md5=ab7366644b11236292820785d074d3dcCAS |
[45] H. A. Patel, S. H. Je, J. Park, D. P. Chen, Y. Jung, C. T. Yavuz, A. Coskun, Nat. Commun. 2013, 4, 1357.
| Crossref | GoogleScholarGoogle Scholar |
[46] H. E. Gottlieb, V. Kotlyar, A. Nudelman, J. Org. Chem. 1997, 62, 7512.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXmvVClsbo%3D&md5=a879555e06b6b3719d3ac47cf5d7570cCAS |
[47] B. Priewisch, K. Rück-Braun, J. Org. Chem. 2005, 70, 2350.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1yisLc%3D&md5=1f71930aa9f29331d903e54dcb1670a9CAS |
[48] B. H. Toby, R. B. Von Dreele, J. Appl. Cryst. 2013, 46, 544.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXjvFWnu7c%3D&md5=b60c1cf5e119253c9ac58882d35b698aCAS |
[49] H. M. D. Bandara, S. Cawley, J. A. Gascón, S. C. Burdette, Eur. J. Org. Chem. 2011, 2916.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmtlaqu7Y%3D&md5=27bc82e28211736a99f0a0f5f964f166CAS |
[50] C. M. Stuart, R. R. Frontiera, R. A. Mathies, J. Phys. Chem. A 2007, 111, 12072.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXht1Kit77J&md5=177e250291d058a69f253d81ecba5c87CAS |
[51] B. K. Pathem, Y. B. Zheng, J. L. Payton, T.-B. Song, B.-C. Yu, J. M. Tour, Y. Yang, L. Jensen, P. S. Weiss, J. Phys. Chem. Lett. 2012, 3, 2388.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtFyqt7nN&md5=5477df874ee30b4287b40b089931bf4aCAS |
[52] Y. B. Zheng, J. L. Payton, C.-H. Chung, R. Liu, S. Cheunkar, B. K. Pathem, Y. Yang, L. Jensen, P. S. Weiss, Nano Lett. 2011, 11, 3447.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXovFKltbg%3D&md5=7aebb869ee2e532181bbc3d3f818f697CAS |
[53] Y. C. Ou, D. S. Zhi, W. T. Liu, Z. P. Ni, M. L. Tong, Chem. – Eur. J. 2012, 18, 7357.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xms1ektL8%3D&md5=025e31e08efd71683486791377487395CAS |
[54] G. A. Senchyk, A. B. Lysenko, H. Krautscheid, E. B. Rusanov, A. N. Chernega, K. W. Krämer, S.-X. Liu, S. Decurtins, K. V. Domasevitch, Inorg. Chem. 2013, 52, 863.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXktV2qtQ%3D%3D&md5=4eb7e9c71dbe96e5b097d836ae880ae1CAS |
[55] A. Spek, J. Appl. Cryst. 2003, 36, 7.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXltlChtw%3D%3D&md5=a70469aed31b7683d455883cff6e2596CAS |
[56] O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard, H. Puschmann, J. Appl. Cryst. 2009, 42, 339.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjsFSnsbg%3D&md5=152b232aadeaa73225d2029119611dc4CAS |
* Associate Professor Deanna D’Alessandro is the recipient of the 2017 Australian Academy of Science Le Févre Prize and the 2017 RACI Alan Sargeson Lectureship (Inorganic Division).