

Predicting the future of plant breeding: complementing empirical evaluation with genetic prediction
Mark Cooper A D , Carlos D. Messina A , Dean Podlich B , L. Radu Totir B , Andrew Baumgarten B , Neil J. Hausmann B , Deanne Wright B and Geoffrey Graham CA DuPont-Pioneer, 7250 NW 62nd Avenue, PO Box 552, Johnston, IA 50131, USA.
B DuPont-Pioneer, 8305 NW 62nd Avenue, PO Box 7060, Johnston, IA 50131, USA.
C DuPont-Pioneer, 7300 NW 62nd Avenue, PO Box 1004, Johnston, IA 50131, USA.
D Corresponding author. Email: mark.cooper@pioneer.com
Crop and Pasture Science 65(4) 311-336 https://doi.org/10.1071/CP14007
Submitted: 4 January 2014 Accepted: 27 February 2014 Published: 23 April 2014
Abstract
For the foreseeable future, plant breeding methodology will continue to unfold as a practical application of the scaling of quantitative biology. These efforts to increase the effective scale of breeding programs will focus on the immediate and long-term needs of society. The foundations of the quantitative dimension will be integration of quantitative genetics, statistics, gene-to-phenotype knowledge of traits embedded within crop growth and development models. The integration will be enabled by advances in quantitative genetics methodology and computer simulation. The foundations of the biology dimension will be integrated experimental and functional gene-to-phenotype modelling approaches that advance our understanding of functional germplasm diversity, and gene-to-phenotype trait relationships for the native and transgenic variation utilised in agricultural crops. The trait genetic knowledge created will span scales of biology, extending from molecular genetics to multi-trait phenotypes embedded within evolving genotype–environment systems. The outcomes sought and successes achieved by plant breeding will be measured in terms of sustainable improvements in agricultural production of food, feed, fibre, biofuels and other desirable plant products that meet the needs of society. In this review, examples will be drawn primarily from our experience gained through commercial maize breeding. Implications for other crops, in both the private and public sectors, will be discussed.
Additional keywords: envirotyping, genetics, genotyping, modeling, phenotyping, physiology, prediction, selection.
Introduction
There is always interest in understanding the major environmental, socioeconomic and emerging scientific trends expected to impact and shape the likely paths that will unfold in the future of global agriculture (Evans 1998; Smith et al. 2005a; Tuberosa et al. 2005; Edgerton 2009; Boyer et al. 2013; Grassini et al. 2013). The need for long-term planning in plant breeding is a given. However, speculating about potential future directions of any scientific activity is always challenging. Here we attempt to chart some intermediate ground between setting some challenging targets to aim for over the next 25 years, while grounding the proposed future possibilities based on emerging, promising research trends.
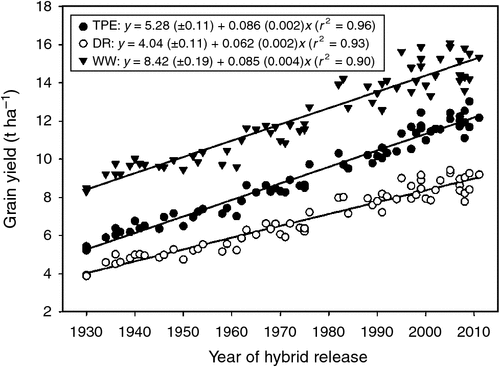
Prior to embarking on such ‘grounded’ speculations about the future of plant breeding, it is instructive to consider a few key lessons that have been learned about genetics and plant breeding over the last Century. First, if we consider the long-term genetic improvement of yield for the major crops, plant breeding has worked whenever there is genetic variation within the germplasm pools accessible to plant breeders and selection has focussed on the right traits measured in the right environments (Allard 1960; Fehr 1984; Hallauer and Miranda Filho 1988; Evans 1996; Cooper and Hammer 1996; Tuberosa et al. 2005). For example, long-term genetic gain for yield of maize in the US has been well documented (Duvick et al. 2004; Fig. 1). Second, the path from research discoveries to impact at the level of the agricultural system can be tortuous and takes time and long-term commitment. Even when a retrospective view of the outcome indicates substantial and rapid progress, the multiple research paths that were explored and the balance of successes and failures encountered along the way are often hidden from full view. Reviews and historical interpretations typically focus on the successful outcomes without equal attention to all of the failures and the important lessons that were learned from these failures. Third, integration across disciplines has always been a hallmark of successful plant breeding. The scientific community that enables plant breeding is large and is built on an integrated network of scientific information and experience that has co-evolved with the discipline of plant breeding (Sprague and Dudley 1988; Lamkey and Lee 2006).
|
Determining the physiological and genetic contributions to realised, long-term yield improvement of crops is complicated. Predicting the limits of sustainable crop yields and enabling future breeding trajectories that move us closer to the potential, sustainable crop yields is even more challenging, but of great importance for society. Duvick et al. (2004) hypothesised that the historical, long-term genetic improvement of maize for the US corn-belt (Fig. 1) has been achieved by combining multiple plant mechanisms to improve tolerance to the stresses that occur in the different environments of the US corn-belt. These agricultural environments are an outcome of the biophysical conditions of the environment (soil, climate and biota) and the crop management practices of the farmers. There is considerable heterogeneity of environmental conditions across the US corn-belt (Löffler et al. 2005). Further, the components of the environment are subject to change with time. Maize farmers continually seek crop management strategies that will provide high and economical yields for their given soil and climate conditions. Thus, the genotype–environment systems of agriculture, within which the breeding programs operate, are not static but are an evolving and moving target for the plant breeder.
Duvick et al. (2004) emphasised the interplay of improved crop management and genetics in the realisation of the long-term genetic gain for yield of maize in the US corn-belt. For example, the trend of US farmers to increase maize plant populations to achieve higher grain yields was enabled through a combination of increased use of nitrogen fertiliser, mechanisation and the development of hybrids that were adapted to the higher plant populations (Duvick et al. 2004; Hammer et al. 2009; Mansfield and Mumm 2014). Access to sufficient water to support the increased biomass and yield demands of the higher plant populations was enabled through use of irrigation where rainfall was not sufficient. Hammer et al. (2009) used a combination of experimental and simulation results to demonstrate that the increase in maize yield in the US corn-belt (Fig. 1) was supported by coordinated genetic improvements in root system architecture and function that enabled improved capacity to access soil water and changes in canopy system architecture that improved radiation use efficiency. This interplay of genetic improvement and optimisation of crop management practices continues today.
The integration of traits for insect protection and herbicide protection into maize hybrids used across the US corn-belt has provided farmers with new options for managing and removing many of the yield-reducing effects caused by insect damage and losses of water through weed competition, both important components of the environmental biota. This widespread change in the farming systems used across the US corn-belt has contributed to the increased grain yields achieved by farmers since the mid-1990s (Fig. 1; Duvick et al. 2004). The widespread deployment of hybrids with insect and herbicide protection has initiated a shift in the emphasis of further genetic improvement and management strategies for effective use of the water and nitrogen resources for the US corn-belt. This shift has further emphasised the importance of genotype × management interactions as an important component of genotype × environment (G×E) interactions. As the genetic improvement of maize for drought tolerance and nitrogen-use efficiency becomes an increasingly important target for breeding programs, increased investment into the co-development of germplasm and resource-efficient management strategies is anticipated over the coming decades.
We will draw on examples from our experience with commercial maize breeding for the US corn-belt. Figure 2 is a schematic of the cyclical process followed in a typical, commercial maize breeding program. The commercial product of the breeding program is a single-cross hybrid (Fig. 2a). Germplasm is improved within two heterotic groups; one is designated as the female pool, often referred to as Stiff-Stalks (SS), and the other is the male pool, often referred to as non-Stiff-Stalks (NSS). New inbreds are developed within each pool and hybrids are created by combining inbreds from the different heterotic groups. Figure 2a distinguishes the key components and stages of the breeding program: (i) sampling of germplasm within the accessible germplasm pool, organised into complementary heterotic groups; (ii) creating new populations of inbreds, thus sampling the possible recombinants that can be generated from the available genetic diversity within the populations; (iii) evaluation of the inbreds in hybrid combinations by choosing suitable testers from the complementary heterotic group; (iv) recycling of the superior new inbreds; and (v) creation, testing and advancement of new hybrid combinations by bringing together improved inbreds from the complementary heterotic groups. Figure 2b identifies the key steps that occur within each stage of the breeding program: (i) obtain a relevant sample of the elite germplasm under evaluation in the stage; (ii) evaluate the sampled germplasm in a relevant sample of environments in specifically designed multi-environment trials (METs); (iii) analyse the trait data collected on the germplasm within the environments in the METs; (iv) make selection decisions based on analysed results obtained from the METs; (v) advance the selected germplasm to the next stage of the breeding program; (vi) make relevant predictions of the expected genetic value and phenotypic performance of the advanced germplasm; and (vii) recycle the improved germplasm and use it to initiate subsequent cycles of the breeding program. The details depicted in Fig. 2b provide additional specifics of the processes involved in the germplasm recycling arrows depicted in Fig. 2a.

|
Looking towards the next 25 years, we can consider some key questions currently being tackled by maize breeders and some plausible paths for the evolution of maize breeding methodology. We will use examples from the stages depicted on the schematic in Fig. 2 to illustrate the opportunities we predict. Two major trends are proposed. First, the scale of commercial maize breeding programs will continue to increase. Second, improved understanding of the genetic architecture of traits, combined with balanced use of native and transgenic sources of genetic diversity, will enhance the breeder’s ability to utilise a range of model-based prediction methodologies to support the increased scale of the breeding programs. We can anticipate that as the cost of molecular technologies continues to decrease these two trends will also apply to other crops and other global geographies. We can also anticipate that the scale of phenotyping capacity, the measurement of the right traits in the right environments, will become a major limitation to implementation of prediction methodologies and as such will be an active area of research focus in the private and public sectors.
Trend 1. Increase in scale of breeding programs
Some of the key successes of plant breeding in the 20th Century originated from large breeding programs that were able to mount long-term efforts (e.g. Wang et al. 2003; Duvick et al. 2004; Borlaug and Dowswell 2005; Kush 2005). Germplasm diversity, trait and genetic knowledge, and an understanding of the target population of environments (TPE) where the products of the breeding programs would be grown were all important in each case. In combination with germplasm knowledge, each of these programs can be characterised as adopting a large ‘numbers game’ breeding strategy; i.e. large numbers of breeding populations were created and large numbers of progeny from the populations were tested in a large number of field plots; field testing was conducted across many locations and years to sample the relevant environmental conditions and management practices of the TPE. For maize breeding in the US corn-belt, the increase in scale was enabled by the development of specialised field equipment to plant, harvest and measure traits from large numbers of experimental plots conducted in the farmers’ fields that would ultimately grow the products of the breeding programs. An interesting feature of these large breeding programs is that they were not typically run as one large centralised breeding program. Instead they operated more like an interconnected network of smaller breeding programs with multiple breeders applying distributed field testing to solve their local problems and exchanging improved germplasm (Fig. 3; Smith et al. 2006). Therefore, you could superimpose the schematic depicted in Fig. 2 multiple times on the distributed network of breeding programs depicted in Fig. 3. This interconnected network of breeding programs introduces a degree of diversity of problem solving at the local level that is arguably important in the technology and germplasm innovations that have contributed to the sustained, long-term genetic gains, both locally and globally (Fig. 1). Podlich and Cooper (1999) used simulation to argue the case that such a large distributed, interconnected network of breeding programs would have advantages over a single large, centralised breeding program. While there is no experimental comparison to test the hypothesis, both scale (large size) and structure (distributed networks of coordinated breeding efforts) appear to be important features of breeding programs for the long-term genetic improvement of yield and other complex traits of the three major crops maize, wheat and rice.

|
While the scaling of breeding programs in the 20th Century was driven by methods to expand field-testing capacity and improve the quality of trait phenotypic data obtained from field experiments, we argue that the scaling of breeding programs in the 21st Century will come from the integrated use of germplasm knowledge, high-throughput genotyping capabilities, improved phenotyping capacity, and the development and deployment of modelling and prediction methods. Components of this integration towards genetic prediction are illustrated below.
Trend 2. Greater use of modelling and prediction methodology
The concept of prediction has a long history in plant breeding. The origins and development of quantitative genetics theory for both plant and animal breeding were strongly motivated by the need to provide a predictive genetic framework to guide the design of breeding methodology (Hill 2014; Walsh 2014). The ‘Breeder’s Equation’ (Lynch and Walsh 1998; Walsh 2005) has been extended into many forms to represent the expected genetic gain from cycle to cycle for alternative breeding methods (e.g. Hallauer and Miranda Filho 1988; Comstock 1996). Recent extensions that combine both theoretical and simulation methods have incorporated information from quantitative trait loci (QTLs) (Lande and Thompson 1990; Dekkers and Chakraborty 2001; Podlich et al. 2004; Cooper et al. 2005). Although these equations can be applied to predict changes in population means over one or a few cycles of recurrent selection, they do not enable the breeder to predict the performance of individual genotypes within the populations in any breeding cycle. An opportunity that emerges from the combined use of high-throughput genotyping and phenotyping methodologies to enable mapping of trait genetic architecture is the ability to predict the expected genetic value of individual genotypes based on their genetic fingerprint (Meuwissen et al. 2001; Podlich et al. 2004; Cooper et al. 2005; Sebastian et al. 2010; Messina et al. 2011).
Below we discuss advances in four key areas that will underpin the proposed trends of increased breeding program scale and utilisation of prediction methods: (1) improved phenotyping methodology, (2) extending germplasm knowledge to the sequence level, (3) trait genetic knowledge, and (4) statistical methodology and information management (IM) systems to enable prediction.
Trend 2.1. Towards improved phenotyping methodology
The process of empirical evaluation of trait phenotypes for genotypes created and advanced through the stages of a breeding program has remained largely unchanged over much of the history of plant breeding. Different versions of Fig. 2, with more or less detail, can be found throughout the plant breeding literature (e.g. Hallauer and Miranda Filho 1988; Fehr 1991; Cooper and Hammer 1996). Typically, the numbers of genotypes the breeder has to evaluate changes by orders of magnitude across stages of the breeding program. Figure 4 represents an alternative view of the schematic depicted in Fig. 2a, in this case focusing on the change in numbers of genotypes at the different stages of the breeding cycle. For purposes of discussion we consider a breeding program that begins each empirical field testing cycle with 104 new genotypes (‘Tested’, Fig. 4). We will return to discuss the ‘Untested’ layer of the cycle below. In the schematic, the initial set of 104 genotypes is reduced by three orders of magnitude to around 101 (Fig. 4) during the course of evaluation and selection (Fig. 2a). The quantity of phenotypic information for each genotype changes with stage of the breeding program. At the early stages with 104 genotypes, evaluation of individual genotypes is based on METs that sample a small number of environment–management combinations from the TPE. In the later stages of the breeding program, the number of genotypes has been reduced by selection and the number of environment–management combinations sampled in the METs increases for the remaining genotypes. Thus, the general pattern over a cycle of a breeding program is that the quantity of phenotypic information increases for the genotypes that advance to the later stages. This increase in quantity of phenotypic information per genotype is indicated by the expanding triangle to the right of Fig. 4. Breeders are interested in any opportunities to increase throughput and improve the quality of trait phenotyping at all stages of the breeding program (Cooper and Hammer 1996; Araus and Cairns 2014). We discuss some promising opportunities.

|
Improved phenotyping methodology: experimental design and analysis
There have been many advances in the design and conduct of plant breeding experiments to reduce the undesirable impact of the different sources of environmental and measurement variation that can occur within the spatial footprint of the experiments. Benefits have been demonstrated from understanding the potential sources of such environmental variation (e.g. soil variability, systematic effects of equipment operation within the experiment) and experimental design and analysis methodology. We consider some examples below.
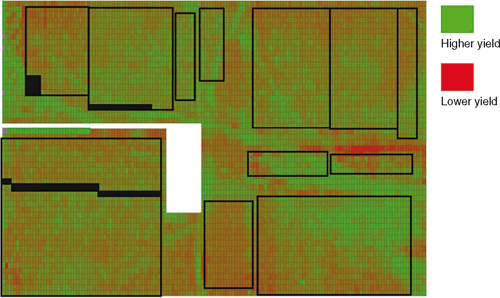
Precision agriculture technologies can be used to create a spatial description of field heterogeneity, thus informing the positioning of experiments within relatively uniform management zones (Fig. 5). Even with effective placement of experiments to avoid previously characterised spatial variation for soil properties, there are still sources of environmental variation that cannot be characterised a priori. There is a long history of statistical research into appropriate experimental designs and analysis procedures for the conduct and interpretation of plant breeding experiments (DeLacy et al. 1996). Although benefits from advanced experimental designs and analyses are realised across all stages of the breeding program, the gains are likely to be greatest in the early stages due to the lower levels of replication used. Advantages have been demonstrated from the use of incomplete block experimental designs that enable adjustment for inter-block variation (Basford et al. 1996; Williams et al. 2002; Piepho and Williams 2006; Williams et al. 2006). These incomplete block designs are often implemented as augmented designs, where test genotypes are included only once per location and check genotypes are replicated (Federer et al. 1975; Federer et al. 2001). Cullis et al. (2006) extended the use of the augmented design to incorporate replication on a specified percentage of the test genotypes.
|
Advances in statistical software, such as ASReml (Gilmour et al. 2009), have enabled the development and application of mixed linear models to account for non-random sources of field variation as well as genetic and environmental covariances commonly found in plant breeding METs. Gilmour et al. (1997) described a systematic process for the adjustment of global and local environmental trends that arise in the conduct of METs, such as those depicted for one field location in Fig. 5. Qiao et al. (2000, 2004) demonstrated the positive impact of combining incomplete-block experimental designs (Williams et al. 2002, 2006) and spatial adjustment procedures (Gilmour et al. 1997) for comparing and selecting genotypes in a wheat breeding program. They demonstrated that the statistical adjustment methods were more effective for predicting the relative yield of genotypes in future years than analyses that did not adjust the data for spatial heterogeneity within the experiments.
Here results are shown to demonstrate further the positive impact that spatial analysis adjustments can have, using grain yield, grain moisture, time to flowering (growing degree units from planting to pollen shed, SHDGDU) and ear height data from a large number of maize experiments conducted in the US corn-Belt (Fig. 6). Following the approach of Gilmour et al. (1997), statistical analysis procedures were applied to account for extraneous variation through random row and column effects and local trend through the two-dimensional, separable autoregressive process of order 1 for the covariance structure of the residuals for neighbouring plots in the row and column directions (AR1 × AR1). The impact of the model adjustment procedure was examined in terms of the change in the genetic correlation between all pairs of locations included within a MET, comparing the correlation coefficients based on unadjusted raw data v. spatially adjusted data. Positive shifts in the distributions of the genetic correlation coefficients obtained from the use of adjusted data were observed for the four traits (Fig. 6). For example, in the case of grain yield (Fig. 6a), based on all pairs of environments from 1126 METs, the average genetic correlation coefficient based on the raw data was 0.25, whereas for the spatially adjusted data, the average correlation coefficient was increased to 0.34. Consistent with the study by Qiao et al. (2000, 2004) for wheat in Australia, applying the framework of Gilmour et al. (1997) to the maize experiments in the US corn-belt, we interpret the positive impact of the statistical adjustment procedures on the genetic correlation coefficient estimates as an improved characterisation of the relative yield performance of the genotypes within the experiments.

|
A natural extension of the single-site analyses used to generate the summary depicted in Fig. 6 is the application of across-environment analyses suitable for METs. Across-environment analyses can be defined to incorporate appropriate covariance structures for G×E interactions. Smith et al. (2001, 2002, 2005b) and van Eeuwijk et al. (2001) describe approaches for modelling the genetic correlations between environments. The simplest covariance structure is compound symmetry, which assumes that all environments have the same genetic variance, and all pairs of environments have the same genetic covariance (and correlation). The uniform-correlation, heterogeneous variance-covariance model fits separate genetic variance components for each environment, but still assumes that each pair of environments has the same genetic correlation. An unstructured covariance model fully relaxes these assumptions by allowing each environment to have a separate genetic variance and each pair of environments to have a separate genetic correlation. An alternative is the factor analytic (e.g. Smith et al. 2001) model, which fits separate genetic variance for each environment and models the covariances between environments as a linear function of one or more factors.
With the increasing availability of software systems that provide the flexibility to implement mixed models that include the within-environment spatial models in combination with experimental design information and appropriate models of G×E interactions, this mixed model framework has become the recommended approach for the analysis of METs (Fig. 2).
Improved phenotyping methodology: managed environments
Comstock (1977) introduced the concept of the ‘target population of environments’ for a breeding program. He defined the TPE to represent the expected mixture of environmental conditions that can be encountered across multiple years within a defined geography. The TPE is the geographical and temporal set of environments within which the breeder is creating and selecting improved genotypes to perform. The different environmental conditions result from the different combinations of soil physical conditions within farm fields, the different management (e.g. planting date, fertiliser levels, irrigation quantity and timing) decisions taken by farmers, and the different weather conditions from year to year. These variable environmental conditions within the TPE influence the outcomes of selection decisions taken by the breeder when they give rise to G×E interactions that change the rank order of the genotypes across the environments for yield and important agronomic traits.
For METs at any stage of the breeding program, the breeder is dealing with a finite sample of environments that sample a relatively small subset of the potential geographical and crop-management conditions in a limited number of years (Fig. 2). The breeder seeks to conduct a MET for each stage of the breeding program that predicts expected relative trait performance of the experimental genotypes in the TPE, particularly as it unfolds in the immediate future years. The presence of G×E interactions, in combination with the sampling variance associated with conducting METs within the context of a TPE, generally slows the rate of realised genetic gain achieved by a breeding program (Podlich et al. 1999). Many strategies have been suggested to deal with G×E interactions and their impact on genetic gain in plant breeding.
Here we introduce the general term ‘envirotyping’ to refer to the collective body of methodologies that are applied to characterise environments within METs and the TPE. The term envirotyping is used here as a complement to the more familiar terms genotyping and phenotyping. Envirotyping the environments (location–year–management combinations) sampled within a MET to assess how they represent the TPE has been widely advocated (e.g. Cooper and Hammer 1996; Fig. 2). However, to do this in practice, within a timeframe that supports the breeder to make informed decisions (e.g. Podlich et al. 1999), requires significant effort beyond the standard plant and harvest steps followed in the conduct of METs. Following the envirotyping methodology introduced by Muchow et al. (1996), Chapman et al. (2000) combined historical weather records with soil survey data and used a crop growth model to develop a drought perspective of the sorghum TPE for north-eastern Australia. They discussed the challenges that a sorghum breeder in north-eastern Australia faces when breeding for future years, given the conduct of a MET in a small sample of the preceding years. Subsequent work has applied similar procedures to develop a view of the TPE for wheat in north-eastern Australia (Chenu et al. 2011). The same methods have been applied for maize in the US corn-belt (Fig. 7). As in the sorghum and wheat studies, the maize case study demonstrates that it is possible to identify typical temporal modes of environmental variation for the soil–plant water balance (Fig. 7a) and characterise their associated spatial patterns for a region (Fig. 7b).

|
A complicating factor for breeders, highlighted in the case studies for all three crops (sorghum, wheat and maize), is that any of the identified temporal patterns of water balance can occur in any location–year combination, albeit with different expected frequencies of occurrence. Thus, the low predictability of any given water-balance pattern (Fig. 7a) at a given location–year combination (Fig. 7b) makes it difficult to determine a priori the traits and magnitude of genetic variation that will be revealed at each location–year combination. Therefore, as important as it is to identify these patterns and the associated genetic variation for relevant traits at the level of the TPE (Chapman et al. 2000; Chenu et al. 2011; Fig. 7), for practical application of this information it is necessary to diagnose, visualise and quantify these in real-time for the location–year combinations sampled in any MET to enable informed selection decisions (Podlich et al. 1999).
Löffler et al. (2005) extended the envirotyping views of the US corn-belt TPE to incorporate biotic and abiotic stresses into the classification system. They developed tools to visualise and quantify the inter-annual variability for the geographical distribution of different environmental conditions (Fig. 8). Considering the time span 2009–2012, the widespread geographical distribution of drought conditions (classified as Temperate Dry) across the US corn-belt in 2012 (Fig. 8a; Boyer et al. 2013) can be observed and compared with the more restricted distribution of drought for the period 2009–2011 (Fig. 8b–d). The availability of such environmental characterisation information to position the conditions sampled at multiple locations in individual years enables the breeder to consider options for weighting the observed genetic variation based on its expected relevance for future years (Podlich et al. 1999).

|
In all three examples (Chapman et al. 2000; Löffler et al. 2005; Chenu et al. 2011; Figs 7, 8), the challenges associated with conducting METs in a finite number of years to predict genotype trait performance in the TPE, or at least the following sequence of years, are emphasised. Field-based, managed-environment methods have been advocated and utilised to enable genetic gain for important environmental components of the TPE that contribute to repeatable G×E interactions (Fischer et al. 1989; Cooper et al. 1995, 1997, 2014; Campos et al. 2004, 2006; Kirigwi et al. 2004; Trethowan et al. 2005; Bänziger et al. 2006; Weber et al. 2012; Rebetzke et al. 2013). Two maize breeding examples and the managed-environment solutions that have been implemented for the US corn-belt TPE to deal with important environmental components and the associated G×E interactions are considered below: brittle snap resistance, and drought resistance.
Managed environments: brittle resistance
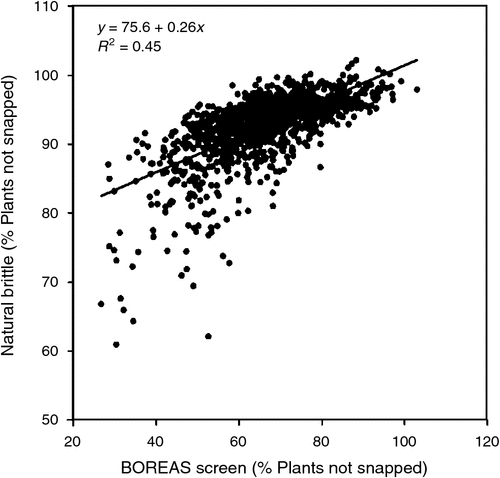
The trait brittle snap in maize occurs when severe wind gusts generated from thunderstorms break the stalks of maize plants during the pre-flowering development stage. The incidence of thunderstorms is identified here as an important environmental component of the US corn-belt TPE. Brittle snap is a concern primarily in the western region of the US corn-belt. One per cent stalk breakage approximately equates to 1% yield loss, since snapped plants do not produce ears and there is little potential for surrounding non-snapped plants to compensate for the snapped plants. There is genetic variation for resistance to brittle snap among maize hybrids. Phenotypic screening for brittle snap is difficult because natural brittle snap events, where appropriate storms coincide with the location of an experiment at the appropriate stage of crop development, are rare and even when they occur they tend to show a high level of spatial variability within a location due to the spatial variability of the gusting effects of the storms. This environmental variability causes the heritability of the brittle snap trait to be extremely low, making it difficult to characterise and select for resistance within a maize breeding program when relying entirely on thunderstorms inducing natural brittle events within experiments. In response to variation for brittle susceptibility among commercial hybrids and the stochastic nature of natural brittle snap events, in the last decade Pioneer has dedicated research to understanding the genetic architecture of brittle resistance and developing screening methodology to select for brittle snap resistance. This research has resulted in the development of wind machines (Boreas machines, named as such after the Greek god of the North wind) and a brittle testing network. The Boreas machines are capable of simulating thunderstorm conditions favourable for inducing brittle snap. Equally important has been the development of expertise in the key environmental and physiological conditions needed to maximise genetic differences for brittle snap resistance among genotypes. Genetic variation for brittle snap resistance under Boreas wind machine testing is normally distributed, with higher heritability than found within natural brittle snap events. Furthermore, positive correlations are seen between Boreas wind machine data and data obtained from informative natural brittle snap events (Fig. 9). The development of an effective, managed-environment approach for brittle snap testing has enabled selection against brittle snap susceptibility that is predictive of the hybrid variation resulting from thunderstorms within the TPE. Thus, a component of the overall G×E interaction for yield of maize in the TPE can be targeted as a breeding objective. This represents an example of the local innovation and problem solving that occurs in a subset of the total set of breeding programs that is subsequently shared and utilised by other breeding programs (Fig. 3).
|
Managed environments: drought resistance
Pioneer has a long history of developing maize hybrids with drought tolerance (Barker et al. 2005; Fig. 1). The incidence of drought is identified here as an important component of the US corn-belt TPE (e.g. Figs 7, 8). Historically, the improvements in hybrid drought tolerance have relied heavily on wide-area testing throughout the relevant geographies of the US corn-belt (Cooper et al. 2006; Fig. 2). It is well recognised that inter-annual rainfall across the US corn-belt can result in a highly variable distribution of drought conditions across years (Löffler et al. 2005; Figs 7, 8). In some years, there can be widespread drought and in other years there can be widespread high rainfall. This variability makes it difficult to ensure, through wide-area testing, adequate testing of new hybrids under appropriate drought conditions every year and for all stages of the breeding program. One approach that has been advocated is the use of drought managed-environments (Barker et al. 2005). This involves working at locations where there is a low likelihood of untimely rainfall, uniform soil over sufficiently large areas to conduct breeding experiments, and access to precision irrigation capacity. When these and other conditions can be achieved, the METs for any stage of a breeding program can be designed and irrigation water inputs can be managed to impose a level of water deficit predictive of important drought conditions in the TPE (Fig. 10; Cooper et al. 2014). The utilisation of drought managed-environment methodology has been implemented by Pioneer for drought maize breeding in the US corn-belt (Barker et al. 2005; Cooper et al. 2014). As with the brittle snap example, the use of managed environments for drought testing has enabled selection for drought tolerance that is predictive of the hybrid variation observed from naturally occurring drought conditions in the western region of the US corn-belt (Cooper et al. 2014).

|
The initial drought breeding effort began in the highly drought-prone, western region of the US corn-belt. However, as shown in Figs 7, 8, drought is also prevalent in other regions of the corn-belt. The germplasm developed through the drought breeding efforts in the Western region and the use of the drought managed-environment technologies is now widely utilised across the US corn-belt. This represents another example of innovation and problem solving in part of the network of breeding programs that has been shared to provide germplasm and technology solutions for other breeding programs and regions throughout the US corn-belt (Fig. 3).
Improved phenotyping methodology: crop growth and development framework
Precision phenotyping of key traits in managed environments (Figs 9, 10) and in METs conducted to sample locations, management and years that are representative of the TPE (Fig. 2b) plays a critical role in enabling effective selection decisions. A key requirement of any test environment is to enable measurement of relevant traits at the appropriate scale necessary for genotype evaluation at the different stages of the breeding program (Fig. 4). Development of new phenotyping technologies that enable quantification of relevant traits previously only noted or scored by breeders enables new opportunities for prediction, improved screening and evaluation of product concepts (Figs 9, 10).
At each stage of product development, phenotyping resources are prioritised towards traits with potential to contribute to yield and agronomic improvement within the TPE, as noted for the brittle snap (Fig. 9) and drought (Fig. 10) examples described above. Phenotyping is informed by envirotyping studies that define the array of potential biotic and abiotic challenges to be faced by the product over its commercial lifecycle (Löffler et al. 2005; Figs 7, 8). In addition, quantitative biological frameworks enabled by a suitable model of crop growth and development can be leveraged to guide phenotyping (Messina et al. 2009). Used appropriately, these frameworks provide insights into the physiological processes that underpin genetic variation for yield and long-term genetic improvement of yield (Fig. 1). The application of a phenotyping strategy guided by such a framework enables evaluation of the merit of genotypes for enhanced resource capture (e.g. radiation, water, nitrogen), resource utilisation efficiency, potential yield sink size and reproductive resilience to stress. With this additional level of phenotyping, traditional empirical selection objectives can be extended to advance material through the breeding program, contributing different modes of action for stress tolerance and for yield potential. The potential benefits of adopting this approach include an increase in functional genetic diversity and the recombination of different modes of action into new genotypes at faster rates than would otherwise be likely (Cooper et al. 2002, 2009; Chapman et al. 2003; Hammer et al. 2005; Messina et al. 2011). Moreover, the efficiency and potential rate of gain of the breeding program can be enhanced through early elimination of germplasm exhibiting undesirable phenotypes that have not been revealed by empirical evaluation in METs.
The application of such a framework to product development requires the development of high-throughput phenotyping methods tailored to multi-tiered phenotyping strategies (Sinclair 2011). Often, quantification of physiological traits that affect crop adaptation to target environments requires complex and expensive phenotyping. Sinclair (2011) advocates the use of relatively simple, high-throughput screens during early stages of product development, with incrementally increasingly detailed phenotyping as successful genotypes progress through later stages of evaluation. Such an approach is consistent with the traditional increase in scale of phenotyping for each genotype with more advanced stages of breeding (Fig. 4) and enables application of selection pressure for multiple physiological modes of action contributing to performance in the TPE.
For example, as applied to breeding maize for improved yield stability under drought stress, this strategy could involve initial phenotyping for anthesis-to-silking interval (ASI), followed by quantification of plant-to-plant variability for yield within plots and final elucidation of dynamic responses of silk emergence to variations in ear growth (Fig. 11). Reductions in ASI at the plot level can be realised through multiple mechanisms, including increased resource capture (Hammer et al. 2009; Messina et al. 2009), biomass allocation to the ear (Vega et al. 2001) and ear growth per ovule (Edmeades et al. 1993). The goal of the first phenotypic screen is to eliminate weak germplasm with critical deficiencies by utilising field traits that are relatively simple and cost-effective. A second-tier screen, based on high-throughput phenotyping of plant-to-plant variability for yield, utilises phenotyping methods that are increasingly complex and more costly. This second screen enables selection of genotypes that have both reduced ASI and improved within-plot plant-to-plant stability to maintain kernel set under stress. Nevertheless, only a subset of the genotypes that meet these criteria will have the desired, improved reproductive efficiency. A final characterisation of silk numbers as a function of ear growth informs the selection of the final few genotypes to advance for further evaluation and for use as new parents for subsequent cycles of breeding (Fig. 2). At this stage, detailed and involved phenotyping is required to describe the functional physiological processes underlying the yield advantage. Figure 11 illustrates a three-tiered phenotyping approach that led to the selection of a hybrid with superior reproductive efficiency relative to a drought-susceptible control. Messina et al. (2009, 2011) used simulation to demonstrate the contribution of reproductive efficiency (Fig. 11) to superior performance of maize hybrids grown under drought-stress conditions and how the relevance of this trait changes with environmental conditions and crop management. Imaging technologies, such as those used to phenotype kernels per ear and silk numbers (Fig. 11d, e) will be increasingly relevant to enable high-throughput phenotyping.

|
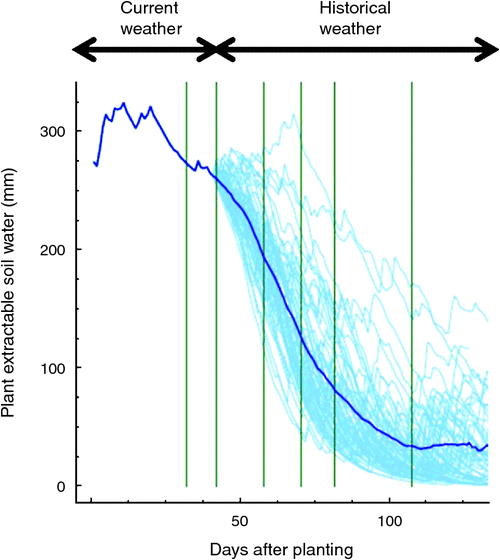
As with the brittle example above, an important aspect of precision phenotyping for drought is the agronomic manipulation of the environment to expose genetic variation for the physiological processes underpinning the traits of interest. Timing of agronomic activities within the field experiments is critical to reveal genotypic differences for adaptive traits. For example, if the goal of the breeder is to improve drought tolerance by increasing reproductive efficiency (Fig. 11), applying irrigation too early or too late to a field experiment can lead to total reproductive failure or over-vigorous growth and reproduction, both outcomes limiting the identification of superior genotypes. Real-time modelling technologies are instrumental to monitor and predict the consequences of crop management on performance and genetic variance for the traits of interest. Figure 12 provides an example of the soil water available to a maize crop from planting to day 45, based on empirical measurement up to that day, and projected soil moisture from day 45 to maturity, based on 60 years of weather data given the management defined for the experiment and the starting conditions, initiated from the empirical measurements up to day 45. Access to such dynamic characterisation of soil water status, and predictions of the likely future changes in water balance for the environments in the MET, enable the breeder to make informed real-time decisions on appropriate irrigation management to increase the likelihood of revealing important trait and yield genetic variation.
|
Trend 2.2. Extending germplasm knowledge to the sequence level
As described above, successful crop improvement involves the breeder working germplasm to create and identify new genotypes that demonstrate improved yield, stress and agronomic performance in the environments of the TPE. Germplasm is the fundamental resource that is manipulated by plant breeders to achieve genetic improvement of target traits. The genetic basis of trait architecture and inheritance underlies the value of the germplasm for genetic improvement (Fig. 1). The improvements are achieved by breeders using their understanding of trait genetic variation within the context of an understanding of the TPE and the available germplasm diversity to create new genetic combinations with improved trait phenotypes for target environments. The new combinations are created by applying complementary breeding methodologies to manipulate the available germplasm resources to realise the potential of the germplasm. The new genotypes so created must be stable in seed production systems that multiply the new genotypes to the appropriate scale for use by the target farmers. The maize heterotic groups utilised today to enable commercial production of single-cross hybrids in North America represent an example of breeders developing and applying a breeding methodology that complemented the potential identified from an understanding of the germplasm resource and the genetic architecture of traits in maize. The maize heterotic group structures and the genetic potential for trait improvement that they enable did not exist a priori to be discovered; rather, they were created through multiple breeding cycles involving generations of plant breeders in the public and private sectors.
Breeders who can effectively characterise their germplasm and relate this characterisation to the genetic architecture of traits are well positioned to understand the factors that have contributed to the success of important genotypes (Fig. 1) and to chart a course for sustained genetic improvements to product performance (Feng et al. 2006; Smith et al. 2006; Messina et al. 2011). Pioneer has a long history of corn breeding in North America and a strong understanding of the pedigree relationships that have contributed to successful products over many decades (Duvick et al. 2004; Smith et al. 2004). With the advent of molecular technologies, Pioneer is not only able to define the key inbreds in the pedigree history but also characterise the fragments of the corn genome that have contributed to their success (Fig. 13; Feng et al. 2006). Pioneer’s corn germplasm universe (Fig. 13a) shows a cross-section of the history of breeding at Pioneer in terms of pedigree relationships dating back to the inbreds of the 1920s. Each successful inbred is represented as a single node and the successful breeding crosses that created improved inbreds are represented as lines connecting nodes. The universe is structured into two heterotic groups (left and right sides), with early founder inbreds in the centre of the universe. The different decades of breeding radiate out from the centre of the pedigree universe, with current elite inbreds on the outer bounds of the universe diagram. This graphic (Fig. 13a) provides another view of the heterotic groups of the germplasm pool discussed in relation to Fig. 2. The pedigree trajectory of one of the current elite inbreds is highlighted (Fig. 13a). The ancestry of this inbred can also be drawn in terms of a traditional pedigree diagram (Fig. 13b).

|
With molecular marker technologies and high-throughput genotyping, breeders can construct whole-genome DNA fingerprints of inbreds and assemble segments of the genome into different haplotypes, representing the genetic diversity embedded within the germplasm. Here haplotypes are defined as alternative versions (i.e. ‘types’) of contiguous sections of a chromosome and as such can be treated as alleles of regions of the genome. Genotypic information can be combined with pedigree information to characterise haplotypes in terms of founder segments, referred to as identity-by-descent (IBD) information (Fig. 13c). The translation of DNA fingerprints for genotypes into IBD information provides a genotypic view of how the genetic diversity available within the founding germplasm has been shaped over cycles of breeding to create the elite germplasm used by breeders today. For example, Fig. 13b shows a trace of the inheritance of a 10 cM region in the maize genome, where the colours represent the haplotypes of founder segments that have been passed down and recombined over generations. When applied to a set of elite inbreds, IBD provides a characterisation of the standing diversity within the elite germplasm and defines the bounds of genetic variation that is being worked within elite × elite breeding crosses (Fig. 13c). New sources of genetic diversity added to the germplasm pool at any stage in the breeding process can be accommodated as new founders and thus introduced into the IBD views.
Estimates of trait effects obtained from mapping studies can be assigned to the different founder haplotypes (e.g. Boer et al. 2007; van Eeuwijk et al. 2010; ter Braak et al. 2010; Bink et al. 2012). The relative value of different founder haplotypes for economically important traits can influence selection decisions. Selection for different haplotypes, either indirectly through selection on phenotype or directly through selection for genetic fingerprint that represents the haplotype, can result in a change in frequency of different haplotypes in the germplasm over time (e.g. Fig. 13d). For example, in some segments of the genome associated with a trait effect, a single founder haplotype can increase in frequency over several decades (e.g. QTL 1, Fig. 13d). In other segments of the genome, the founder haplotype frequencies can remain largely unchanged despite association with trait effects (e.g. QTL 2, Fig. 13d). The latter example can occur when founder haplotypes have competing effects on multiple traits (e.g. positive for yield and negative for grain moisture content) or where QTL × environment interactions occur (e.g. Boer et al. 2007). Trait effects characterised in terms of founder haplotypes provide a framework to define prediction targets in terms of individual QTL regions and for multiple QTLs across the whole genome. With the availability of high-throughput genotyping capabilities, it is possible to characterise new, untested individuals in terms of founder haplotypes and provide qualitative and quantitative predictions of trait performance for all genotypes within a breeding program. These predictions can be used to frontload the set of recombinants that are evaluated in the field, and thus increase the likelihood of developing superior products. Through such implementations of genetic prediction, the effective scale of a breeding program can be increased without the necessity to scale the phenotyping requirement and empirical footprint of all stages of the breeding program (Fig. 4). An additional layer of genotypes that are evaluated based only on their genetic fingerprint (e.g. Fig. 13c) and associated trait effects but are ‘untested’ directly for trait phenotypes in METs or any experimental conditions increases the effective scale of the breeding program without increasing the scale of the empirical phenotyping components.
Trend 2.3. Expanding trait genetic knowledge
Much of our current knowledge of trait genetic architecture in crop plants comes from mapping traits in populations that were specifically designed to identify QTLs (e.g. Boer et al. 2007). Some QTLs of strong effect have been refined to the level of the functional sequence polymorphism (e.g. Salvi et al. 2007) and subsequently used as a component of a dynamic developmental model (Dong et al. 2012). In contrast to the majority of the public-sector efforts, private-sector breeding programs have focussed on developing mapping methods that can be applied to the elite populations and experiments generated at different stages of the breeding program (Figs 2–4). Thus, in addition to mapping and selection within specific crosses (e.g. Boer et al. 2007), methods have been developed for mapping within multi-parent, multi-cross, pedigree-related mating designs that are common within pedigree breeding programs (van Eeuwijk et al. 2010; ter Braak et al. 2010; Bink et al. 2012). Mapping within such multi-cross mating designs has been enabled through utilisation of IBD information to connect haplotype diversity across multiple, pedigree-related segregating populations that are generated, tested and phenotyped during the course of conducting the cycles of the breeding program in the TPE (Figs 2, 13). The effects of the alleles of QTLs identified in such reference populations can be readily related to the haplotype effects segregating in the elite populations of genotypes advancing through the stages of the breeding program (Figs 2–4, 13). Thus, genetic prediction at the levels of parent selection, cross creation, population selection, and individual inbred and hybrid selection within the breeding program cycle are now all feasible (Fig. 2). Quantitative methodologies for implementing this prediction framework are discussed below.
Trend 2.4. Enabling prediction through use of whole-genome evaluation techniques
The fourth key area that underpins the trend of greater use of modelling and prediction is the ability to perform accurate genetic evaluation of the candidates for selection in all stages of the breeding program. While the quantitative machinery of whole-genome prediction methodology may appear as a black box to many, it is, in principle, a direct application of quantitative genetics enabled by the availability of high-throughput genotyping, mixed model statistical methodology and high-performance computing hardware to exercise the algorithms defined in terms of quantitative genetic models (Walsh 2014). Here we review some key points.
Traditional genetic evaluation techniques (Henderson 1984; Hallauer and Miranda Filho 1988; Hill 2014) rely exclusively on the use of phenotypic and pedigree data to estimate dataset-specific genetic parameters that are then used within Henderson’s mixed model equations (HMME) to obtain best linear unbiased predictors (BLUP) for the candidates for selection. Both Likelihood and Bayesian statistical methods have been developed to perform this type of genetic evaluation (Sorensen and Gianola 2002). As illustrated by the ‘Breeder’s Equation’, the expected increase in response to selection per generation depends on the accuracy of the BLUP estimates, the intensity of selection applied by the breeder, and the genetic variability expressed for the trait of interest in the dataset/population under investigation (Lynch and Walsh 1998; Hill 2014). Although highly effective in terms of realised genetic gain for some traits, the traditional phenotypic and pedigree-based genetic evaluation approaches have limited utility for some traits and stages of the breeding process (Dekkers and Hospital 2002).
The advent of cost-effective molecular marker systems, such as single nucleotide polymorphisms (SNPs), has created the opportunity to introduce a new source of information in the routine genetic evaluation process. As illustrated in Fig. 13, these types of data can be used to trace the inheritance of specific chromosomal segments in extended pedigrees through IBD probability computations and thus allow the classification of germplasm at the DNA level. Initial attempts to incorporate the newly classified DNA data within the routine genetic evaluation process have focussed on a two-step approach. First, statistical genetics analysis techniques—linkage and/or linkage disequilibrium (association) mapping—are used to identify QTLs, defined here as variable-size DNA-segment polymorphisms associated with a measurable impact on phenotypic traits of interest. Second, QTLs deemed ‘statistically significant’ at an agreed-upon threshold are fitted as fixed or random terms in a modified version of HMME and used to generate marker assisted BLUPs (MA-BLUPs) for the candidates for selection (Fernando and Grossman 1989; Lande and Thompson 1990). This approach is commonly referred to as marker-assisted selection (MAS). As discussed by Dekkers and Hospital (2002), the use of MAS for practical breeding purposes can be especially challenging for complex traits where inheritance is controlled by a large number of QTLs with small effects. For example, in active breeding programs it is the norm rather than the exception to have traits determined by a large number of QTLs, each with a small effect (e.g. van Eeuwijk et al. 2010). Figure 14a shows the cumulative distribution of the genetic variance generated by a typical mode of inheritance for a trait that is under selection in an active maize breeding program. Note that to explain 100% of genetic variance estimated for this trait, the cumulative effect of >300 cM of the whole maize genome has to be accounted for. For example, a MAS improvement program that is focused on the top 10 chromosomal regions explaining variability for this trait would account for <20% of the total estimated genetic variance.

|
To overcome this problem, Meuwissen et al. (2001) proposed an alternative, marker-based genetic evaluation approach. They advocated using random regression BLUP or model-averaging Bayesian Markov Chain Monte Carlo techniques that fit simultaneously in the statistical model all SNPs available, thus eliminating the need to pre-screen SNPs based on agreed-upon statistical significance thresholds. The underlying assumption behind this approach is that the joint probability distribution between allele states (reflected in linkage disequilibrium measures) and, respectively, between allele origins (reflected in co-segregation measures) at any two loci (observed SNP and unobserved causative locus) can be best exploited by fitting explicitly in the statistical model the ‘contribution’ that each SNP scored on the candidate for selection has on its own expressed phenotype, regardless of the size of this ‘contribution’ (Fig. 14a). Note that while Meuwissen et al. (2001) have developed and discussed the genomic selection concepts using primarily SNP markers, this approach is directly applicable for other types of genetic marker loci used to define haplotypes. The whole-genome evaluation (genomic selection) approach introduced by Meuwissen et al. (2001) has the advantage that, while remaining a black box from a biological understanding viewpoint, it allows the breeder to utilise the entire genetic variability captured by the statistical model even if the true genetic architecture of a trait remains unknown (Fig. 14a, Cooper et al. 2006; Podlich et al. 2004).
Many scientific publications have been written in recent years on both the statistical aspects of the methodology used to implement genomic selection (Gianola et al. 2006; Piepho 2009; Habier et al. 2011) and the utility and potential practical implications of using this novel genetic evaluation technique (Heffner et al. 2009). Empirical evaluations relevant to plant breeding have been summarised (Crossa et al. 2014) and potential extensions beyond the basic additive genetic model have been proposed (Heslot et al. 2013; Marjoram et al. 2014).
From an operational standpoint, implementing a molecular-marker based, whole-genome evaluation program requires careful development of relevant estimation (training) phenotypic datasets. These are then used to estimate the ‘contribution’ to the total genetic variability expressed of each segregating SNP scored on the pool of genomes that form a given estimation dataset (Fig. 14a). Once each SNP has been assigned its own ‘contribution’ by performing the whole-genome statistical analysis of choice on a given estimation dataset, the expected genetic value of any new individual can be obtained by assigning to, and then the summing over, the estimated effects of its own SNP fingerprint. However, it is important to recognise that whole-genome evaluations generated using this approach are specific to the estimation datasets used. To illustrate this point, Fig. 14b shows the decrease in predictive ability for three distinct evaluation datasets whose members have their expected genetic value predicted using a marker-based, whole-genome evaluation procedure trained on a common estimation dataset. Results are shown for nine traits of different heritability collected for F1 hybrids grown in 2 years, 2007 and 2008. Predictive ability represents the correlation between predictions (constructed based on marker data and 2007 phenotypic data) and actual observed phenotypic performance in the field in 2008. Only phenotypic data from 2007 were used to create the whole-genome predictions for the F1 hybrids grown in 2008. Fig. 14b, Real_97 refers to 97 F1 hybrids grown in both 2007 and 2008, where the correlation between the actual field performances of these 97 F1 hybrids across years is used as an empirical benchmark. Correlation coefficients between predicted and observed trait phenotypes for three categories of hybrids were tested in 2008: Tested × Tested (T×T), Tested × Untested (T×U) and Untested × Untested (U×U). In Fig. 14b, T×T_495 means that both inbred parents of 495 F1 hybrids grown only in 2008 also had other progeny with phenotypic data in 2007; T×U_595 means that one of the two inbred parents of 595 F1 hybrids grown only in 2008 did not have any progeny with phenotypic data in 2007; U×U_153 means that both inbred parents of 153 F1 hybrids did not have any progeny with phenotypic data in 2007. As expected, a decrease in predictive ability occurs once the members of the evaluation set become more distinct from the reference population that forms the estimation set used to assign individual SNP ‘contributions’, which are then used to create the predictions of the new individuals; the correlation coefficients typically follow the trend Real > T×T > T×U > U×U. Differences in the predictive ability for traits of the same heritability could indicate the presence of G×E interactions contributing to the unknown components of the trait genetic architecture (Cooper et al. 2006). This example illustrates the performance of one estimation dataset when applied to predict the expected genetic value for members of three evaluation datasets from within the same stage of the breeding program. However, a commercial breeding program generates thousands of potential estimation datasets in each growing season. Significant effort has to be put into optimising the use of field data for estimation-set design to maximise predictive ability and information management systems to support the prediction process.
From the perspective of commercial or public breeding programs, the availability of molecular-marker based, whole-genome evaluation techniques, as discussed above, creates the opportunity for breeders to accurately predict the expected genetic value of genotypes in all stages of a breeding program: inbred parent selection, breeding cross design, segregating population kernel selection, double haploid (DH) and/or recombinant inbred line (RIL) evaluation and advancement to future inbred parent status, and hybrid creation, selection and characterisation. However, this requires developing, implementing and taking full advantage of the complete spectrum of enabling technologies needed to increase genetic gain, including high-throughput genotyping, appropriate phenotyping technologies and phenotyping capacity as discussed above, information management systems, data analysis and visualisation capabilities. If all enabling technologies are in place, it is possible to develop integrated breeding systems that optimally use both field-based breeding programs and molecular–virtual based breeding programs to maximise genetic gain with optimal use of resources. It must be recognised, however, that although field experimental design and analysis is a well-established and mature area, molecular-marker based experimental (estimation set) design and analysis is a new research area that needs significant additional research work.
Combining the phenotyping methods discussed above with germplasm knowledge and high-density genotyping has enabled genetic prediction for complex traits for the elite germplasm of a breeding program. For both brittle snap and drought, consistent high-quality phenotypic data have allowed detection of QTLs and design of training datasets for whole-genome prediction for improved agronomic and yield performance. These QTLs and training datasets are currently being utilised within the Pioneer maize breeding program through MAS and whole-genome prediction approaches (e.g. Fig. 15; Cooper et al. 2014).

|
For purposes of demonstration, two traits that are relevant to breeding for drought performance are discussed further. For the two quantitative traits growing degree units from planting to pollen shed (SHDGDU) (Fig. 15a, c) and for the anthesis-to-silking interval (ASIGDU) (Fig. 15b, d), a linear association is typically observed between whole-genome predictions and phenotypes observed in independent experiments for inbreds sampled from both the SS and NSS heterotic groups. In this example the inbreds are evaluated as hybrids, with appropriate testers selected from the complementary heterotic group, as depicted in Fig. 2a. The presence of such linear associations between whole-genome predictions and independently observed trait phenotypes (Fig. 15) indicates that the genetic effects of the haplotypes estimated in the training datasets are predictive of the effects of the same haplotypes when these are present in the hybrids evaluated in the independent target experiments. In this case, the genetic relationship between the hybrids in the training datasets and the target experiments is a consequence of the pedigree relationships between the hybrids comprising the datasets compared (Fig. 13). The linear associations between the predictions and independent observations can deviate from 1 : 1 relationships (Fig. 15), indicating differences between the trait genetics expressed in the two datasets. However, the linear associations are suitable to enable ranking and selection of inbreds, based on hybrid performance, on the whole-genome predictions (Fig. 15).
Germplasm: maintaining and expanding access to functional genetic diversity
The commercial maize breeder of today works with elite germplasm that has been shaped over multiple cycles of breeding by generations of maize breeders (Figs 1–3, 13; Duvick et al. 2004; Smith et al. 2006; Feng et al. 2006). The founding, open-pollinated populations and the inbred lines that were created over the history of the breeding program (Fig. 13a) have been preserved in cold storage. This germplasm legacy can be genotyped and phenotyped to study how breeding has shaped germplasm diversity over time (Figs 1, 13; Duvick et al. 2004; Feng et al. 2006). From the mid-1990s, maize hybrids that were commercialised in the US corn-belt began to incorporate transgenes for insect protection and herbicide protection. The transition to utilisation of transgenes for these traits was rapid. In the first decade of the 21st Century, commercial maize hybrids were predominantly designed as products based on improved elite native germplasm including one or more transgenes incorporated through a backcrossing strategy. This trend has continued, and in the 2010s, commercial maize hybrids used in the US now incorporate multiple transgenes for protection against different insects and multiple herbicides. Utilisation of transgenes for insect and herbicide protection is an example of breeding programs seeking novel functional genetic diversity outside that available in the elite germplasm pools. Maize breeders will continue to seek such exotic functional genetic diversity from outside the elite germplasm pools whenever this improves the performance of the hybrids that can be developed. Insect and herbicide resistance will continue to be relevant trait targets for new sources of genetic diversity. In addition, novel sources of disease resistance and abiotic stress tolerance will be areas of focus over the next 25 years. There are many challenges associated with the discovery of transgenes conferring efficacious tolerances for abiotic stresses (Passioura 2006, 2012). Aligning the functional basis of high-throughput phenotyping conducted through use of pot experiments in controlled environment facilities with relevant field-based targets is an essential component of any such efforts. Effective, field-based phenotyping of the candidate genes within the elite germplasm of the target crop in the relevant environments of the TPE will be a critical component to determine the product potential of these novel sources of genetic diversity. Some illustrative and encouraging results are appearing (e.g. Nelson et al. 2007; Castiglioni et al. 2008; Guo et al. 2013; Habben et al. 2014).
Predicting product concepts: crop growth and development modelling
Predicting performance of maize hybrids for a complex, diverse and continually evolving TPE, such as the US corn-belt, is a long-term challenge for breeders. Genotype × environment × management (G×E×M) interactions are ubiquitous (Messina et al. 2009). As discussed above, maize breeders have had success with this inference problem (Fig. 1) by evaluating hybrids for yield, agronomics, and biotic and abiotic stress tolerance in METs that sample multiple years and locations that cover the geographical area of the TPE (Fig. 2). The sampling of environments in METs is an attempt to capture repeatable aspects of the environment and management variation encountered in on-farm production conditions throughout the TPE (Figs 2, 7–11). Augmenting traditional phenotypic selection with genetic predictions for traits can be used to increase the scale of breeding programs to further deal with the inference challenge (Figs 4, 14, 15). Yet, the stochasticity and diversity of environments in an evolving TPE places limits on our capabilities to make accurate predictions (Figs 7, 8; Podlich et al. 1999). This challenge to enabling prediction for breeding is emphasised above in the need for research into methods for design and creation of appropriate estimation datasets to train the genetic prediction models. Crop growth and development models structured to explicitly capture variation for the biophysical processes that determine yield and agronomic trait variation can be used to augment and extend the accuracy of genetic predictions for hybrid performance.
Crop growth and development models are structured on biophysical principles that encapsulate resource capture and use-efficiency concepts (Passioura 1977). These models provide a quantitative biological framework for harnessing genotypic, environmental, management and physiological knowledge to enable predictions of hybrid performance (Cooper et al. 2002; Hammer et al. 2005, 2006; Messina et al. 2009). The analysis of these predictions offers the potential to leverage repeatable components of G×E×M interactions at any given stage of the breeding process. Realising this potential requires estimation of parameters in process equations within the crop growth and development framework that are unique to a genotype, making predictions unique for this entity (Cooper et al. 2009; Messina et al. 2011). Within this framework, the challenge of making inferences of hybrid performance based on field testing (Fig. 2) shifts to the challenge of modelling and phenotyping physiological processes to enable accurate predictions of the norm of reaction of hybrids for key traits across the environmental conditions of the TPE, or at least repeatable components of the TPE (e.g. Figs 9, 10). Recent advances in our understanding of the environments of the TPE, maize physiology, phenotyping technologies, and execution of experiments in managed stress environments (e.g. Figs 10, 11) have enabled refinement of the models and implementation of phenotyping strategies with enough resolution to produce predictions applicable to the large number of genotype and environment combinations necessary to support the plant-breeding advancement process (Fig. 4; Messina et al. 2011; Cooper et al. 2014). The predictions obtained from simulation of trait norms of reaction for a TPE, based on these crop models, augment the empirical datasets obtained from METs and enable additional evaluation of the hybrids to a scale greater than could be performed using only the empirical data obtained from METs.
An example following the methodology described by Messina et al. (2011) is used to illustrate the simulation of the norms of reaction for a set of 10 maize hybrids (Fig. 16) in the final stage of a selection cycle (Fig. 4). Extending the fitness/adaptation landscape models of Wright (1932) and Kauffman (1993), Cooper et al. (2005) defined the yield-response surface for a reference pool of germplasm in the context of a TPE. Within this theoretical framework, the norm of reaction of any genotype is defined as the collection of yield values for all relevant environmental combinations that can occur in the TPE. Useful views of hybrid norms of reaction can be constructed by focusing on hybrid performance in the key environmental conditions within the TPE identified by comprehensive envirotyping (e.g. Figs 7, 8). To visualise the yield norm of reaction, Messina et al. (2011) introduced the two-dimensional yield–trait performance landscape, where the genotypic values for a trait genetic model are ordered on the horizontal axis and the yield values are ordered on the vertical axis (Fig. 16a). These yield–trait performance landscapes can be constructed for the key environment-types identified by envirotyping (Fig. 7; Messina et al. 2011). For each genotype class, given an appropriate ordering for the genetic model of the trait of interest, represented on the horizontal axis, the yield distribution resulting from variation for all other traits is represented as a density profile on the vertical axis, indicated in Fig. 16a by different colours.

|
Messina et al. (2011) used the yield–trait performance landscape (Fig. 16a) to examine the expected relationship between traits and yield within a reference germplasm pool, represented by the solid black line, for drought and favourable environment-types identified for the US corn-belt by envirotyping. Further, they used the graphic to project expected trajectories over cycles of selection for a breeding program, represented by the blue line (Fig. 16a). Within any cycle of the breeding program, individual genotypes can be represented by positions on the graphic, indicated by black points (Fig. 16a), determined by their trait genotype and yield values.
In addition to viewing the positions of individual genotypes on the yield–trait performance landscape for specific environmental conditions (Fig. 16a), many different representations of the norm of reaction of the genotypes are possible. For example, for 10 genotypes (hybrids), their locations on the yield–trait performance landscape can be defined for one environment-type revealed by envirotyping (Fig. 16a), or their yield for different environments within a given year can be displayed on a geographical grid (Fig. 16b). The relationship between the two views of the norms of reaction for the hybrids in a TPE is revealed through the environmental characterisation of individual locations (e.g. Fig. 12), the relationship of the individual locations to the environment-types (Fig. 7) and their organisation on a geographical grid for the chosen year (Fig. 16b) or for multiple years. An advantage of the geographical display depicted in Fig. 16b is that this prediction view of the norm of reaction is similar to the typical location–year format of plant-breeding METs.
Here Fig. 16b provides examples of yield predictions for a set of 10 hybrids, representative of the final, pre-commercial stage of a breeding program (Fig. 4), evaluated in a grid of ~6000 soil–weather environment combinations that occurred across the US corn-belt in 2012. Many of these simulated environments were not encountered or sampled in prior stages of testing in traditional METs (Fig. 2), or through empirical testing in 2012, and the predictions exposed potential strengths and weaknesses of the 10 candidate commercial hybrids that otherwise would most likely only have surfaced in future production conditions, post-commercialisation. In this case, the simulated norms of reaction (Fig. 16b) enabled improved selection decisions at advanced stages of product development, and the simulations are complementary to the empirical results obtained from the METs conducted in 2012.
Similar geographical views or inter-annual views for specific or groups of locations can be generated for any hybrid at any stage of the breeding program (Fig. 4). However, generating such views for every hybrid at all stages of a breeding program is of limited interest when large numbers of hybrids are to be considered. Of greater interest is the creation of graphics and metrics for sorting the hybrids, such as the yield–trait performance landscapes (e.g. Fig. 16a). In the yield–trait performance landscape, every genotype has a relative position on the landscape graphic based on its predicted yield and trait values. Thus, the geographical views of each of the 10 hybrids shown in Fig. 16b represent specific projections of yield on a geographical grid that each directly map to a position on the yield–trait fitness landscape shown in Fig. 16a. Given that the predictions of hybrid performance depicted in Fig. 16 are based on process-level phenotypes, we propose referring to this approach as ‘phenotypic prediction’. As the underlying crop growth model and the environmental inputs used to generate the yield predictions are continually improved, graphical views, such as those shown in Fig. 16, provide opportunities for the breeder to make inferences about the expected norms of reaction of individual and groups of hybrids across current and potential future conditions of the TPE.
The parameters of the crop growth models that are estimated for the hybrids, to enable the phenotypic predictions shown in Fig. 16, can themselves be treated as trait phenotypes of the hybrids. As such, the genetic variation for these model parameters can be mapped like any other trait. By mapping the genetic architecture of the model parameters, it is possible to make a phenotypic prediction using the crop growth model for any genetic combination of the model parameters (Messina et al. 2011). Thus, by combining genetic prediction with phenotypic prediction, enabled by the crop growth model, it is possible for the breeder to consider the trait norm of reaction for the hybrids that are advancing through the breeding program and for the untested genotypes that have yet to enter the testing program (Fig. 17). With appropriate genetic and crop models of trait phenotypes, the potential of this approach is that the breeder can pre-select the untested genotypes and frontload the breeding program with new inbreds that have increased likelihood of producing improved hybrids with desirable norms of reaction for the TPE. Thus, the breeding program is a source of both the critical data necessary to create the training datasets used to build the prediction models for the reference germplasm of the breeding program and also the data used to evaluate any predictions that are made about the untested genotypes, once they enter the breeding program and are tested in future stages and cycles of the breeding program.

|
With advances in information-management capabilities, whole-genome prediction methodology (Figs 13–15) and high-throughput computing, application of phenotypic prediction (Figs 16, 17) to untested recombinants and hybrids evaluated at early stages of product development (Fig. 4) is now feasible for individual breeding programs and within reach to scale to multiple breeding programs (Fig. 3). The methodology for scaling the genetic models is built on a framework that partitions the model of performance into predictable and unpredictable components (Cooper et al. 2009). Within this framework the crop model accounts for a fraction of the predictable component by integrating whole-genome predictions at the process level for traits and capturing repeatable features of the environment-types created by the physiological characteristics of the genotype. In the example presented in Fig. 16, individual hybrids created in a breeding program are projected in a yield–trait performance landscape (Messina et al. 2011). Each point along the yield axis results from a summary across environments that can be decomposed into performance in the TPE for any given year, e.g. Fig. 16b for 2012. It is important to note that the simulated performance of the genotypes is driven by the physiological and genetic characteristics of the hybrids’ growth and development patterns, as these dynamically influence the observed environmental conditions (and types). Such coupling of crop growth and development models with whole-genome prediction capabilities allows breeders to consider developing products for a TPE that will change in the long term with the evolving germplasm, environmental conditions and changes in crop management (Fig. 1). At early stages of the breeding process, where there are many untested genotypes (Fig. 4), prediction for the new genotypes that have yet to undergo evaluation in METs relies fully on whole-genome prediction applied to the estimation of parameters in the crop growth and development model process equations that characterise the physiology of the crop in the environments of the TPE (Fig. 17). Early results from application of this methodology to support the development of drought-tolerant maize hybrids for the US corn-belt are discussed by Cooper et al. (2014). As with the other prediction methodologies described in this review, we anticipate that understanding the limits to applications of such prediction enabled by crop-growth models will be the focus of research in the coming years.
Discussion
The fundamentals of plant breeding still apply today as they did in the past. For the commercial maize breeder, a working knowledge of germplasm, an understanding of trait genetics and the target population of environments, high-throughput phenotyping and a practical application of selection theory, combined with a clear definition of product targets, will continue to be foundational to successful maize hybrid development. Advances in breeding technologies are allowing us to build on these foundations. We have emphasised two coevolving trends that are anticipated to be features of plant breeding for the foreseeable future: (1) increase in scale of breeding programs, enabled by (2) modelling and use of prediction methodology at all stages of breeding. Both of these trends are already unfolding in commercial maize-breeding programs operating in the US corn-belt. There are already commercial maize hybrids used by farmers in the US corn-belt developed using the methodologies considered in this review (Cooper et al. 2014). We expect these trends to continue in the commercial maize-breeding sector and expand globally over the next 25 years and mature to become increasingly foundational to commercial maize breeding. As the value of the breeding technologies is demonstrated in the major crops and the costs of the technologies decrease, accelerated adoption of applications in other crops is anticipated.
Most of the traits targeted for improvement by the commercial maize breeder have been considered genetically complex. From the results of multiple mapping studies conducted over the last decade, today we have confirmation of this assumption and an empirical understanding of the genetic architecture of the traits for the elite germplasm used in breeding programs (Feng et al. 2006; Boer et al. 2007; van Eeuwijk et al. 2010; Figs 13, 14). For some traits, where a large body of QTL and gene-based information exists across multiple species, such as flowering time, dynamic gene-to-phenotype models have been constructed and predictions from these models have been tested (Dong et al. 2012).
The toolkit of the commercial maize breeder today is different from that of 10 years ago. Directed use of native variation in elite populations through use of marker technologies and incorporation of transgenic sources of genetic diversity for insect and herbicide protection is now commonplace for product targets in the US corn-belt. Molecular technologies now enable detailed views of the maize genome, provide unprecedented access to sequence data, and allow the study of the effects of selection from molecular to whole-plant phenotypic levels. Combining high-throughput genotyping and phenotyping technologies to enable molecular-enhanced predictions of trait performance has opened new ways to evaluate the germplasm worked by the commercial maize breeder (Figs 4, 14–17; Messina et al. 2011).
Prior to the availability of genetic predictions for traits, the maize breeder had to phenotype every individual to obtain any trait assessment of the new genotypes created to initiate a cycle of breeding. Family predictions based on pedigree relationships were possible. However, individuals from within the same family could not be distinguished. This limited the number of individuals that the breeder could work within a breeding program to the scale of the phenotyping that was possible within the resources of the breeding program. For many of the traits of interest, phenotyping requires replication in the appropriate environmental conditions. Some of the traits, such as the brittle snap and drought tolerance examples discussed above, require specialised environmental conditions, equipment and measurement expertise. Such trait phenotyping can be expensive. For a typical maize-breeding program, this phenotyping requirement limits the numbers of individuals that are used to initiate a cycle of breeding (Fig. 4). Today, genotyping of individuals to the level of the linkage disequilibrium that exists within the pedigree-related reference populations of the breeding program (Fig. 13) can be done more cheaply than the phenotyping of all important traits. Therefore, with the enablement of the prediction methodologies discussed here, the breeder can obtain an assessment of many trait phenotypes for an individual before experimental phenotyping. This enables the breeder to increase the scale of the breeding program to numbers of genotypes that are orders of magnitude beyond those that can be directly phenotyped (Fig. 4). The scale of the genotype numbers tested within the breeding program and within the advancement process is depicted as ranging from 104 in the early stages to 101 in the final stages. An additional layer of new genotypes that are not tested directly in METs within the breeding program, but are evaluated by prediction, can now be included as part of the cycle of the breeding program; these are the 105–106 untested genotypes that are characterised by molecular markers and evaluated by prediction (Figs 14, 15). Therefore, the evaluation process in the commercial maize-breeding program today relies heavily on genotyping and prediction in the initial stages to complement and extend empirical phenotyping. As the genotypes enter the breeding program, a phased increase in direct phenotyping begins. By the stage of commercial release, the evaluation of hybrid potential is determined predominantly by direct phenotyping in the commercial environments of the TPE (Figs 2, 4). This empirical evaluation in METs can be further augmented through use of appropriately designed and parameterised crop growth models (Figs 16 and 17; Messina et al. 2009, 2011). In addition, the new inbreds that demonstrate high breeding value are integrated into new cycles of elite germplasm, and in parallel with their use in new commercial hybrids, they are also used as parents in new cross combinations to initiate new cycles of the breeding program (Fig. 2). Further, the inbreds are shared across breeding programs (Fig. 3) and the hybrids are evaluated broadly across the geographical area of the TPE to sustain the long-term genetic gain (Duvick et al. 2004; Fig. 1).
Our prediction for the future of plant breeding, at least for the next 25 years, is that the trends we have discussed here for commercial maize-breeding programs in the US will continue and the scale of commercial maize-breeding programs will increase and there will be increased utilisation of prediction methodologies to enable this increase in scale. We can also anticipate that, as the cost per data point of genotyping and additional molecular technologies continues to decrease, these trends will be adopted for other crops. This expansion to crops beyond maize has already begun in the large, commercial breeding companies (Sebastian et al. 2010). The opportunities created by genetic prediction have re-emphasised the importance of trait phenotyping. Trait phenotyping today not only enables the traditional evaluation and advancement process of the breeding program but also provides the resource for construction of estimation datasets to enable genetic prediction and phenotypic prediction (Fig. 17). The scale of the data resources utilised by the breeder has required complementary advances in the information-management infrastructure to support breeding programs. This information management need will continue as the scale of breeding programs continues to expand.
Trait phenotyping (Fig. 11), envirotyping (Figs 7, 8, 12), genetic (Figs 14, 15) and phenotypic (Figs 16, 17) prediction, and data-management tools have made considerable and often vast amounts of data available to the breeder to support decisions at all stages of the breeding program cycle (Figs 2, 4, 17). Trait phenotyping is evolving to the point where breeders will have at their disposal information that provides insights into physiological determinants of adaptation in the context of the important environmental conditions of the TPE (Messina et al. 2009; Munns et al. 2010; Furbank and Tester 2011). It is anticipated that integrated utilisation of this information can improve rates of genetic gain for important target environments (Hammer et al. 2005; Cooper et al. 2009, 2014; Fig. 10). Dynamic models of crop growth structured around concepts of resource capture and utilisation efficiency will provide a capability to integrate trait information across multiple, non-linear physiological relationships (Hammer et al. 2006; Messina et al. 2011) and guide multi-layered phenotyping strategies (Figs 10, 11, 15) to support product development (Cooper et al. 2014). Integration of the effects of traits on yield via crop models provides a capability to simulate the expected norm of reaction for hybrid yield in the TPE (Fig. 16). Studying the hybrid yield norm of reaction has the potential to provide novel insights about the functional importance of genomic regions will contribute to an improved understanding of the germplasm diversity available to the breeder and how breeding strategies and selection methods can achieve directed changes in the norm of reaction for hybrid yield in the TPE (Cooper et al. 2002, 2009; Hammer et al. 2006; Messina et al. 2011).
The transition from conventional to molecular-enhanced breeding in commercial maize-breeding programs has been rapid and has relied on multiple advances in molecular, genetic, breeding, phenotyping, modelling and informatics technologies (Cooper et al. 2004, 2006; Eathington et al. 2007). These and other technology advances that are not covered in this review are interconnected components of the breeding strategy, and the breeder operates as an integrator of the technologies in the execution of a breeding program strategy with short- and long-term objectives. An important area for consideration is the education and training of the future generation of plant breeders and technology innovators. Collaboration between the commercial-sector breeding community and the universities that provide the formal science education and the initial training of new plant breeders is already happening and is an area for greater attention into the future.
Acknowledgements
The comprehensive research effort that is necessary to develop and advance new breeding methodologies, such as those described here, relies on committed and unselfish team efforts. The authors acknowledge the many coordinated and collaborative team efforts that were undertaken over the last decade within the Pioneer research community to enable the research paths that were explored and the discoveries that were ultimately adopted. Ultimately, success from such efforts is motivated by the opportunity and responsibility to improve the sustainability of global agricultural systems for the benefit and needs of society and future generations.
References
Allard RW (1960) ‘Principles of plant breeding.’ (John Wiley and Sons, Inc.: New York)Araus JL, Cairns JE (2014) Field high-throughput phenotyping: the new crop breeding frontier. Trends in Plant Science 19, 52–61.
| Field high-throughput phenotyping: the new crop breeding frontier.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhs1CrtbjL&md5=0fa17f25347429636c978178a971de1cCAS | 24139902PubMed |
Bänziger M, Setimela PS, Hodson D, Vivek B (2006) Breeding for improved abiotic stress tolerance in maize adapted to southern Africa. Agricultural Water Management 80, 212–224.
| Breeding for improved abiotic stress tolerance in maize adapted to southern Africa.Crossref | GoogleScholarGoogle Scholar |
Barker T, Campos H, Cooper M, Dolan D, Edmeades G, Habben J, Schussler J, Wright D, Zinselmeier C (2005) Improving drought tolerance in maize. Plant Breeding Reviews 25, 173–253.
Basford KE, Williams ER, Cullis BR, Gilmour A (1996) Experimental design and analysis for variety trials. In ‘Plant adaptation and crop improvement’. (Eds M Cooper, GL Hammer) pp. 125–138. (CAB International: Wallingford, UK)
Bink MCAM, Totir LR, ter Braak CJF, Winkler CR, Boer MP, Smith OS (2012) QTL linkage analysis of connected populations using ancestral marker and pedigree information. Theoretical and Applied Genetics 124, 1097–1113.
| QTL linkage analysis of connected populations using ancestral marker and pedigree information.Crossref | GoogleScholarGoogle Scholar |
Boer MP, Wright D, Feng L, Podlich DW, Luo L, Cooper M, van Eeuwijk FA (2007) A mixed-model quantitative trait loci (QTL) analysis for multiple-environment trial data using environmental covariables for QTL-by-environment interactions, with an example in maize. Genetics 177, 1801–1813.
| A mixed-model quantitative trait loci (QTL) analysis for multiple-environment trial data using environmental covariables for QTL-by-environment interactions, with an example in maize.Crossref | GoogleScholarGoogle Scholar | 17947443PubMed |
Borlaug NE, Dowswell CR (2005) Feeding a world of ten billion people: A 21st century challenge. In ‘In the wake of the double helix from the Green Revolution to the Gene Revolution. Proceedings of International Congress’. 27–31 May 2003, University of Bologna, Italy. (Eds R Tuberosa, RL Phillips, M Gale) pp. 3–23. (Avenue Media: Bologna, Italy)
Boyer JS, Byrne P, Cassman KG, Cooper M, Delmer D, Greene T, Gruis F, Habben J, Hausmann N, Kenny N, Lafitte R, Paszkiewicz S, Porter D, Schlegel A, Schussler J, Setter T, Shanahan J, Sharp RE, Vyn TJ, Warner D, Gaffney J (2013) The US drought of 2012 in perspective: A call to action. Global Food Security 2, 139–143.
| The US drought of 2012 in perspective: A call to action.Crossref | GoogleScholarGoogle Scholar |
Campos H, Cooper M, Habben JE, Edmeades GO, Schussler JR (2004) Improving drought tolerance in maize: a view from industry. Field Crops Research 90, 19–34.
| Improving drought tolerance in maize: a view from industry.Crossref | GoogleScholarGoogle Scholar |
Campos H, Cooper M, Edmeades GO, Löffler C, Schussler JR, Ibañez M (2006) Changes in drought tolerance in maize associated with fifty years of breeding for yield in the U.S. corn belt. Maydica 51, 369–381.
Castiglioni P, Warner D, Bensen RJ, Anstrom DC, Harrison J, Stoecker M, Abad M, Kumar G, Salvador S, D’Ordine R, Navarro S, Back S, Fernandes M, Targolli J, Dasgupta S, Bonin C, Luethy MH, Heard JE (2008) Bacterial RNA chaperones confer abiotic stress tolerance in plants and improved grain yield in maize under water-limited conditions. Plant Physiology 147, 446–455.
| Bacterial RNA chaperones confer abiotic stress tolerance in plants and improved grain yield in maize under water-limited conditions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXnsVyhsb4%3D&md5=1534167dce0024f598fc24f10d2757ceCAS | 18524876PubMed |
Chapman SC, Hammer GL, Butler DG, Cooper M (2000) Genotype by environment interactions affecting grain sorghum. III. Temporal sequences and spatial patterns in the target population of environments. Australian Journal of Agricultural Research 51, 223–233.
| Genotype by environment interactions affecting grain sorghum. III. Temporal sequences and spatial patterns in the target population of environments.Crossref | GoogleScholarGoogle Scholar |
Chapman S, Cooper M, Podlich D, Hammer G (2003) Evaluating plant breeding strategies by simulating gene action and dryland environment effects. Agronomy Journal 95, 99–113.
| Evaluating plant breeding strategies by simulating gene action and dryland environment effects.Crossref | GoogleScholarGoogle Scholar |
Chenu K, Cooper M, Hammer GL, Mathews KL, Dreccer MF, Chapman SC (2011) Environment characterization as an aid to wheat improvement: interpreting genotype-environment interactions by modeling water-deficit patterns in north-eastern Australia. Journal of Experimental Botany 62, 1743–1755.
| Environment characterization as an aid to wheat improvement: interpreting genotype-environment interactions by modeling water-deficit patterns in north-eastern Australia.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjsFyis7w%3D&md5=8282c863a9541a9c8d5865e5ced1e870CAS | 21421705PubMed |
Comstock RE (1977) Quantitative genetics and the design of breeding programs. In ‘Proceedings of the International Conference on Quantitative Genetics’. 16–21 August 1976. (Eds E Pollack, O Kempthorne, TB Bailey Jr) pp. 705–718. (Iowa State University Press: Ames, IA, USA)
Comstock RE (1996) ‘Quantitative genetics with special reference to plant and animal breeding.’ (Iowa State University Press: Ames, IA, USA)
Cooper M, Hammer GL (Eds) (1996) ‘Plant adaptation and crop improvement.’ (CAB International: Wallingford, UK)
Cooper M, Woodruff DR, Eisemann RL, Brennan PS, DeLacy IH (1995) A selection strategy to accommodate genotype-by-environment interaction for grain yield of wheat: managed-environments for selection among genotypes. Theoretical and Applied Genetics 90, 492–502.
| A selection strategy to accommodate genotype-by-environment interaction for grain yield of wheat: managed-environments for selection among genotypes.Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DC%2BC2c7htlWkug%3D%3D&md5=4a3098061fbf4918c25e8932ad55f642CAS | 24173943PubMed |
Cooper M, Stucker RE, DeLacy IH, Harch BD (1997) Wheat breeding nurseries, target environments, and indirect selection for grain yield. Crop Science 37, 1168–1176.
| Wheat breeding nurseries, target environments, and indirect selection for grain yield.Crossref | GoogleScholarGoogle Scholar |
Cooper M, Chapman SC, Podlich DW, Hammer GL (2002) The GP problem: Quantifying gene-to-phenotype relationships. In Silico Biology 2, 151–164.
Cooper M, Smith OS, Graham G, Arthur L, Feng L, Podlich DW (2004) Genomics, genetics, and plant breeding: A private sector perspective. Crop Science 44, 1907–1913.
| Genomics, genetics, and plant breeding: A private sector perspective.Crossref | GoogleScholarGoogle Scholar |
Cooper M, Podlich DW, Smith OS (2005) Gene to phenotype models and complex trait genetics. Australian Journal of Agricultural Research 56, 895–918.
| Gene to phenotype models and complex trait genetics.Crossref | GoogleScholarGoogle Scholar |
Cooper M, Smith OS, Merrill RE, Arthur L, Podlich DW, Löffler CM (2006) Integrating breeding tools to generate information for efficient breeding: Past, present, and future. In ‘Plant Breeding: The Arnel R. Hallauer International Symposium’. (Eds KR Lamkey, M Lee) pp. 141–154. (Blackwell Publishing Ltd: Oxford, UK)
Cooper M, van Eeuwijk FA, Hammer GL, Podlich DW, Messina C (2009) Modeling QTL for complex traits: detection and context for plant breeding. Current Opinion in Plant Biology 12, 231–240.
| Modeling QTL for complex traits: detection and context for plant breeding.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjsVOguro%3D&md5=525fdc7554e28e23400f0b09dbc24d71CAS | 19282235PubMed |
Cooper M, Gho C, Leafgren R, Tang T, Messina C (2014) Breeding drought tolerant maize hybrids for the US corn-belt: Discovery to product. Journal of Experimental Botany
| Breeding drought tolerant maize hybrids for the US corn-belt: Discovery to product.Crossref | GoogleScholarGoogle Scholar | 24596174PubMed | in press.
Crossa J, Perez P, Hickey J, Burgueño J, Ornella L, Cerón-Rojas J, Zhang X, Dreisigacker S, Babu R, Li Y, Bonnett D, Mathews K (2014) Genomic prediction in CIMMYT maize and wheat breeding programs. Heredity 112, 48–60.
| Genomic prediction in CIMMYT maize and wheat breeding programs.Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DC%2BC3srjsl2qsg%3D%3D&md5=db9e78d8d18ac60efef3e62c7fce5e60CAS | 23572121PubMed |
Cullis BR, Smith AB, Coombes NE (2006) On the design of early generation variety trials with correlated data. Journal of Agricultural, Biological & Environmental Statistics 11, 381–393.
| On the design of early generation variety trials with correlated data.Crossref | GoogleScholarGoogle Scholar |
Dekkers JC, Chakraborty R (2001) Potential gain for optimizing multigeneration selection on an identified quantitative trait locus. Journal of Animal Science 79, 2975–2990.
Dekkers JCM, Hospital F (2002) The use of molecular genetics in the improvement of agricultural populations. Nature Reviews. Genetics 3, 22–32.
| The use of molecular genetics in the improvement of agricultural populations.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XhsV2gsbw%3D&md5=6345e0c3dbb4b8be81a64fdda57c08dbCAS |
DeLacy IH, Basford KE, Cooper M, Bull JK, McLaren CG (1996) Analysis of multi-environment trials—An historical perspective. In ‘Plant adaptation and crop improvement’. (Eds M Cooper, GL Hammer) pp. 39–124. (CAB International: Wallingford, UK)
Dong Z, Danilevskaya O, Abadie T, Messina C, Coles N, Cooper M (2012) A gene regulatory network model for floral transition of the shoot apex in maize and its dynamic modeling. PLoS ONE 7, e43450
| A gene regulatory network model for floral transition of the shoot apex in maize and its dynamic modeling.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xht1CnsbzL&md5=708b79b9b2151caee74ba74b1a29bf1fCAS | 22912876PubMed |
Duvick DN, Smith JSC, Cooper M (2004) Long-term selection in a commercial hybrid maize breeding program. In ‘Plant Breeding Reviews 24: Long term selection: Crops, animals, and bacteria’. Vol. 24, Part 2. (Ed. J Janick), pp. 109–151. (John Wiley & Sons: New York)
Eathington SR, Crosbie TM, Edwards MD, Reiter RS, Bull JK (2007) Molecular markers in a commercial breeding program. Crop Science 47, S154–S163.
| Molecular markers in a commercial breeding program.Crossref | GoogleScholarGoogle Scholar |
Edgerton MD (2009) Increasing crop productivity to meet global needs for feed, food, and fuel. Plant Physiology 149, 7–13.
| Increasing crop productivity to meet global needs for feed, food, and fuel.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjt1Wqs7g%3D&md5=8a6f1d964c86a1381f0680bb2dad6b27CAS | 19126690PubMed |
Edmeades GO, Bolaños J, Hernandez M, Bello S (1993) Causes for silk delay in lowland tropical maize population. Crop Science 33, 1029–1035.
| Causes for silk delay in lowland tropical maize population.Crossref | GoogleScholarGoogle Scholar |
Evans LT (1996) ‘Crop evolution, adaptation and yield.’ (Cambridge University Press: Cambridge, UK)
Evans LT (1998) ‘Feeding the ten billion: plants and population growth.’ (Cambridge University Press: Cambridge, UK)
Federer WT, Nair RC, Raghavarao D (1975) Some augmented row-column designs. Biometrics 31, 361–374.
| Some augmented row-column designs.Crossref | GoogleScholarGoogle Scholar |
Federer WT, Reynolds M, Crossa J (2001) Combining results from augmented designs over sites. Agronomy Journal 93, 389–395.
| Combining results from augmented designs over sites.Crossref | GoogleScholarGoogle Scholar |
Fehr WR (Ed.) (1984) ‘Genetic contributions to yield gains of five major crop plants.’ CSSA Special Publication No. 7. (American Society of Agronomy and Crop Science Society of America: Madison, WI, USA)
Fehr WR (1991) ‘Principles of cultivar development. Vol. 1, Theory and technique.’ (Macmillan Publishing Company: London)
Feng L, Sebastian S, Smith S, Cooper M (2006) Temporal trends in SSR allele frequencies associated with long-term selection for yield of maize. Maydica 51, 293–300.
Fernando RL, Grossman M (1989) Marker assisted selection using best linear unbiased prediction. Genetics, Selection, Evolution. 21, 467–477.
| Marker assisted selection using best linear unbiased prediction.Crossref | GoogleScholarGoogle Scholar |
Fischer KS, Edmeades GO, Johnson EC (1989) Selection for the improvement of maize yield under moisture-deficits. Field Crops Research 22, 227–243.
| Selection for the improvement of maize yield under moisture-deficits.Crossref | GoogleScholarGoogle Scholar |
Furbank RT, Tester M (2011) Phenomics technologies to relieve the phenotyping bottleneck. Trends in Plant Science 16, 635–644.
| Phenomics technologies to relieve the phenotyping bottleneck.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFOhu7%2FJ&md5=7502fe5daf66f7e17f225d5ce9ad745aCAS | 22074787PubMed |
Gianola D, Fernando RL, Stella A (2006) Genomic-assisted prediction of genetic value with semiparametric procedures. Genetics 173, 1761–1776.
| Genomic-assisted prediction of genetic value with semiparametric procedures.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XptVeiu74%3D&md5=3b1a705db79f975f579ed25070503cc2CAS | 16648593PubMed |
Gilmour AR, Cullis BR, Verbyla AP (1997) Accounting for natural and extraneous variation in the analysis of field experiments. Journal of Agricultural, Biological & Environmental Statistics 2, 269–293.
| Accounting for natural and extraneous variation in the analysis of field experiments.Crossref | GoogleScholarGoogle Scholar |
Gilmour AR, Gogel BJ, Cullis BR, Thompson R (2009) ‘ASReml user guide release 3.0.’ (VSN International Ltd: Hemel Hempstead, UK) Available at: www.vsni.co.uk
Grassini P, Eskridge KM, Cassman KG (2013) Distinguishing between yield advances and yield plateaus in historical crop production trends. Nature Communications 4, 2918
| Distinguishing between yield advances and yield plateaus in historical crop production trends.Crossref | GoogleScholarGoogle Scholar | 24346131PubMed |
Guo M, Rupe MA, Wei J, Winkler C, Goncalves-Butruille M, Weers B, Cerwick S, Dieter JA, Duncan KE, Howard RJ, Hou Z, Löffler CM, Cooper M, Simmons CR (2013) Maize ARGOS1 (ZAR1) transgenic alleles increase hybrid maize yeild. Journal of Experimental Botany
| Maize ARGOS1 (ZAR1) transgenic alleles increase hybrid maize yeild.Crossref | GoogleScholarGoogle Scholar | 24218327PubMed |
Habben JE, Bao X, Bate NJ, DeBruin J, Dolan D, Hasegawa D, Helentjaris TG, Lafitte HR, Lovan N, Mo H, Reimann K, Schussler JR (2014) Transgenic alteration of ethylene biosynthesis increases grain yield in maize under field drought-stress conditions. Plant Biotechnology Journal
| Transgenic alteration of ethylene biosynthesis increases grain yield in maize under field drought-stress conditions.Crossref | GoogleScholarGoogle Scholar | 24618117PubMed | in press.
Habier D, Fernando RL, Kizilkaya K, Garrick DJ (2011) Extension of the Bayesian alphabet for genomic selection. BMC Bioinformatics 12, 186–198.
| Extension of the Bayesian alphabet for genomic selection.Crossref | GoogleScholarGoogle Scholar | 21605355PubMed |
Hallauer AR, Miranda Filho JB (1988) ‘Quantitative genetics in maize breeding.’ 2nd edn. (Iowa State University Press: Ames, IA)
Hammer GL, Chapman S, van Oosterom E, Podlich DW (2005) Trait physiology and crop modelling as a framework to link phenotypic complexity to underlying genetic systems. Australian Journal of Agricultural Research 56, 947–960.
| Trait physiology and crop modelling as a framework to link phenotypic complexity to underlying genetic systems.Crossref | GoogleScholarGoogle Scholar |
Hammer G, Cooper M, Tardieu F, Welch S, Walsh B, van Eeuwijk F, Chapman S, Podlich D (2006) Models for navigating biological complexity in breeding improved crop plants. Trends in Plant Science 11, 587–593.
| Models for navigating biological complexity in breeding improved crop plants.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xht1eqtbzJ&md5=122672c6efb119c2840a8645be9ffde0CAS | 17092764PubMed |
Hammer GL, Dong Z, McLean G, Doherty A, Messina C, Schussler J, Zinselmeier C, Paszkiewicz S, Cooper M (2009) Can changes in canopy and/or root systems architecture explain historical maize yield trends in the U.S. corn belt? Crop Science 49, 299–312.
| Can changes in canopy and/or root systems architecture explain historical maize yield trends in the U.S. corn belt?Crossref | GoogleScholarGoogle Scholar |
Heffner EL, Sorrells ME, Jannink JL (2009) Genomic selection for crop improvement. Crop Science 49, 1–12.
| Genomic selection for crop improvement.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjsF2it78%3D&md5=aecf6a77c7c8c592fde59ef52e072856CAS |
Henderson CR (1984) ‘Applications of linear models in animal breeding.’ (University of Guelph: Guelph, ON, Canada)
Heslot N, Akdemir D, Sorrells ME, Jannink JL (2013) Integrating environmental covariates and crop modeling into the genomic selection framework to predict genotype by environment interactions. Theoretical and Applied Genetics
| Integrating environmental covariates and crop modeling into the genomic selection framework to predict genotype by environment interactions.Crossref | GoogleScholarGoogle Scholar | 24264761PubMed |
Hill WG (2014) Applications of population genetics to animal breeding, from Wright, Fisher and Lush to genomic prediction. Genetics 196, 1–16.
| Applications of population genetics to animal breeding, from Wright, Fisher and Lush to genomic prediction.Crossref | GoogleScholarGoogle Scholar | 24395822PubMed |
Kauffman SA (1993) ‘The origins of order: self-organization and selection in evolution.’ (Oxford University Press: New York)
Kirigwi FM, van Ginkel M, Trethowan R, Sears RG, Rajaram S, Paulsen GM (2004) Evaluation of selection strategies for wheat adaptation across water regimes. Euphytica 135, 361–371.
| Evaluation of selection strategies for wheat adaptation across water regimes.Crossref | GoogleScholarGoogle Scholar |
Kush GS (2005) Green revolution: challenges ahead. In ‘In the wake of the bouble helix from the Green Revolution to the Gene Revolution. Proceedings of an International Congress’. 27–31 May 2003, University of Bologna, Italy. (Eds R Tuberosa, RL Phillips, M Gale) pp. 37–51. (Avenue Media: Bologna, Italy)
Lamkey KR, Lee M (Eds) (2006) ‘Plant breeding: The Arnel R. Hallauer International Symposium.’ (Blackwell Publishing Ltd: Oxford, UK)
Lande R, Thompson R (1990) Efficiency of marker assisted selection in the improvement of quantitative traits. Genetics 124, 743–756.
Löffler CM, Wei J, Fast T, Gogerty J, Langton S, Bergman M, Merrill B, Cooper M (2005) Classification of maize environments using crop simulation and geographic information systems. Crop Science 45, 1708–1716.
| Classification of maize environments using crop simulation and geographic information systems.Crossref | GoogleScholarGoogle Scholar |
Lynch M, Walsh B (1998) ‘Genetics and analysis of quantitative traits.’ (Sinauer Associates, Inc.: Sunderland, MA, USA)
Mansfield BD, Mumm RH (2014) Survey of plant density tolerance in U.S. maize germplasm. Crop Science 54, 157–173.
| Survey of plant density tolerance in U.S. maize germplasm.Crossref | GoogleScholarGoogle Scholar |
Marjoram P, Zubair A, Nuzhdin SV (2014) Post-GWAS: where next? More samples, more SNPs or more biology. Heredity 112, 79–88.
| Post-GWAS: where next? More samples, more SNPs or more biology.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhvFCqs7rL&md5=a2f4af49cad4a9e554a60b9244bce872CAS | 23759726PubMed |
Messina CD, Hammer GL, Dong Z, Podlich D, Cooper M (2009) Modelling crop improvement in a GxExM framework via gene-trait-phenotype relationships. In ‘Crop physiology: interfacing with genetic improvement and agronomy’. (Eds V Sadras, D Calderini) pp. 235–265. (Elsevier: Amsterdam)
Messina CD, Podlich D, Dong Z, Samples M, Cooper M (2011) Yield-trait performance landscapes: from theory to application in breeding maize for drought tolerance. Journal of Experimental Botany 62, 855–868.
| Yield-trait performance landscapes: from theory to application in breeding maize for drought tolerance.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVekurs%3D&md5=d56efdbbb118c508c2d6196cc27dc97dCAS | 21041371PubMed |
Meuwissen TH, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157, 1819–1829.
Muchow RC, Cooper M, Hammer GL (1996) Characterizing environmental challenges using models. In ‘Plant adaptation and crop improvement’. (Eds M Cooper, GL Hammer) pp. 349–364. (CAB International: Wallingford, UK)
Munns R, James RA, Sirault XRR, Furbank RT, Jones HG (2010) New phenotyping methods for screening wheat and barley for beneficial responses to water deficit. Journal of Experimental Botany 61, 3499–3507.
| New phenotyping methods for screening wheat and barley for beneficial responses to water deficit.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtVert7jJ&md5=52df62a3d7c02be06e1fec9ca091326aCAS | 20605897PubMed |
Nelson DE, Repetti PP, Adams TR, Creelman RA, Wu J, Warner DC, Anstrom DC, Bensen RJ, Castiglioni PP, Donnarummo MG, Hinchey BS, Kumimoto RW, Maszle DR, Canales RD, Krolikowski KA, Dotson SB, Gutterson N, Ratcliffe OJ, Heard JE (2007) Plant nuclear factor Y (NF-Y) B subunits confer drought tolerance and lead to improved yields on water-limited acres. Proceedings of the National Academy of Sciences of the United States of America 104, 16450–16455.
| Plant nuclear factor Y (NF-Y) B subunits confer drought tolerance and lead to improved yields on water-limited acres.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXht1Whs7%2FK&md5=d30c145cec1aa275f1a18049dd7ca934CAS | 17923671PubMed |
Passioura JB (1977) Grain yield, harvest index, and water use of wheat. Journal of the Australian Institute of Agricultural Science 43, 117–120.
Passioura JB (2006) The perils of pot experiments. Functional Plant Biology 33, 1075–1079.
| The perils of pot experiments.Crossref | GoogleScholarGoogle Scholar |
Passioura JB (2012) Phenotyping for drought tolerance in grain crops: when is it useful to breeders? Functional Plant Biology 39, 851–859.
| Phenotyping for drought tolerance in grain crops: when is it useful to breeders?Crossref | GoogleScholarGoogle Scholar |
Piepho HP (2009) Ridge regression and extensions for genomewide selection in maize. Crop Science 49, 1165–1176.
| Ridge regression and extensions for genomewide selection in maize.Crossref | GoogleScholarGoogle Scholar |
Piepho HP, Williams ER (2006) A comparison of experimental designs for selection in breeding trials with nested treatment structure. Theoretical and Applied Genetics 113, 1505–1513.
| A comparison of experimental designs for selection in breeding trials with nested treatment structure.Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DC%2BD28nlt1KisQ%3D%3D&md5=8efb2bc03de4d708a11133d8fc04fd06CAS | 17028902PubMed |
Podlich DW, Cooper M (1999) Modelling plant breeding programs as search strategies on a complex response surface. Lecture Notes in Computer Science 1585, 171–178.
| Modelling plant breeding programs as search strategies on a complex response surface.Crossref | GoogleScholarGoogle Scholar |
Podlich DW, Cooper M, Basford KE (1999) Computer simulation of a selection strategy to accommodate genotype–environment interactions in a wheat recurrent selection programme. Plant Breeding 118, 17–28.
| Computer simulation of a selection strategy to accommodate genotype–environment interactions in a wheat recurrent selection programme.Crossref | GoogleScholarGoogle Scholar |
Podlich DW, Winkler CR, Cooper M (2004) Mapping as you go: An effective approach for marker-assisted selection of complex traits. Crop Science 44, 1560–1571.
| Mapping as you go: An effective approach for marker-assisted selection of complex traits.Crossref | GoogleScholarGoogle Scholar |
Qiao CG, Basford KE, Delacy IH, Cooper M (2000) Evaluation of experimental designs and spatial analyses in wheat breeding trials. Theoretical and Applied Genetics 100, 9–16.
| Evaluation of experimental designs and spatial analyses in wheat breeding trials.Crossref | GoogleScholarGoogle Scholar |
Qiao CG, Basford KE, Delacy IH, Cooper M (2004) Advantage of single-trial models for response to selection in wheat breeding multi-environment trials. Theoretical and Applied Genetics 108, 1256–1264.
| Advantage of single-trial models for response to selection in wheat breeding multi-environment trials.Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DC%2BD2c3gvVaitA%3D%3D&md5=053fd0d6860fbf464904e33ffa0c9729CAS | 14689186PubMed |
Rebetzke GJ, Chenu K, Biddulph B, Moeller C, Deery DM, Rattey AR, Bennett D, Barrett-Lennard EG, Mayer JE (2013) A multisite managed environment facility for targeted trait and germplasm phenotyping. Functional Plant Biology 40, 1–13.
| A multisite managed environment facility for targeted trait and germplasm phenotyping.Crossref | GoogleScholarGoogle Scholar |
Salvi S, Sponza G, Morgante M, Tomes D, Niu X, Fengler KA, Meeley R, Ananiev EV, Svitashev S, Bruggemann E, Li B, Hainey CF, Radovic S, Zaina G, Rafalski JA, Tingey SV, Miao GH, Phillips RL, Tuberosa R (2007) Conserved noncoding genomic sequences associated with a flowering-time quantitative trait locus in maize. Proceedings of the National Academy of Sciences of the United States of America 104, 11376–11381.
| Conserved noncoding genomic sequences associated with a flowering-time quantitative trait locus in maize.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXnvFOmsrc%3D&md5=73ceba4981548722531c6a5468cab688CAS | 17595297PubMed |
Sebastian SA, Streit LG, Stephens PA, Thompson JA, Hedges BR, Fabrizius MA, Soper JF, Schmidt DH, Kallem RL, Hinds MA, Feng L, Hoeck JA (2010) Context-specific marker-assisted selection for improved grain yield in elite soybean populations. Crop Science 50, 1196–1206.
| Context-specific marker-assisted selection for improved grain yield in elite soybean populations.Crossref | GoogleScholarGoogle Scholar |
Sinclair TR (2011) Challenges in breeding for yield increase for drought. Trends in Plant Science 16, 289–293.
| Challenges in breeding for yield increase for drought.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnsVyrs74%3D&md5=55a3758d80767b21fac1311f1f0c0f90CAS | 21419688PubMed |
Smith A, Cullis B, Thompson R (2001) Analyzing variety by environment data using multiplicative mixed models and adjustments for spatial field trend. Biometrics 57, 1138–1147.
| Analyzing variety by environment data using multiplicative mixed models and adjustments for spatial field trend.Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DC%2BD38%2Fjs12jsw%3D%3D&md5=9bdddf9e3204620ada215d0846da9981CAS | 11764254PubMed |
Smith A, Cullis B, Thompson R (2002) Exploring variety-environment data using random effects AMMI models with adjustments for spatial field trend: Part 1: Theory. In ‘Quantitative genetics, genomics, and plant breeding’. (Ed. MS Kang) pp. 323–335. (CAB International: Wallingford, UK)
Smith JSC, Duvick DN, Smith OS, Cooper M, Feng L (2004) Chages in pedigree backgrounds of Pioneer brand maize hybrids widely grown from 1930 to 1999. Crop Science 44, 1935–1946.
| Chages in pedigree backgrounds of Pioneer brand maize hybrids widely grown from 1930 to 1999.Crossref | GoogleScholarGoogle Scholar |
Smith JSC, Smith OS, Lamkey KR (2005a) Maize breeding. Maydica 50, 185–192.
Smith AB, Cullis BR, Thompson R (2005b) Centenary Review: The analysis of crop cultivar breeding and evaluation trials: an overview of current mixed model approaches. The Journal of Agricultural Science 143, 449–462.
| Centenary Review: The analysis of crop cultivar breeding and evaluation trials: an overview of current mixed model approaches.Crossref | GoogleScholarGoogle Scholar |
Smith S, Löffler C, Cooper M (2006) Genetic diversity among maize hybrids widely grown in contrasting regional environments in the United States during the 1990s. Maydica 51, 233–242.
Sorensen D, Gianola D (2002) ‘Likelihood, Bayesian, and MCMC methods in quantitative genetics.’ (Springer-Verlag: Berlin, Heidelberg)
Sprague GF, Dudley JW (Eds) (1988) ‘Corn and corn improvement.’ 3rd edn (American Society of Agronomy, Inc., Crop Science Society of America, Inc., Soil Science Society of America, Inc., Publishers: Madison, WI, USA)
ter Braak CJF, Boer MP, Totir LR, Winkler CR, Smith OS, Bink MCAM (2010) Identity-by-descent matrix decomposition using latent ancestral allele models. Genetics 185, 1045–1057.
| Identity-by-descent matrix decomposition using latent ancestral allele models.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFOnu7jP&md5=bcf7a4b74568480554ccdee2d3f1472eCAS |
Trethowan RM, Reynolds MW, Sayre K, Ortiz-Monasterio I (2005) Adapting wheat cultivars to resource conserving farming practices and human nutritional needs. Annals of Applied Biology 146, 405–413.
| Adapting wheat cultivars to resource conserving farming practices and human nutritional needs.Crossref | GoogleScholarGoogle Scholar |
Tuberosa R, Phillips RL, Gale M (Eds) (2005) ‘In the wake of the double helix from the Green Revolution to the Gene Revolution. Proceedings of an International Congress.’ 27–31 May 2003 University of Bologna, Italy. (Avenue Media: Bologna, Italy)
van Eeuwijk FA, Cooper M, DeLacy IH, Ceccarelli S, Grando S (2001) Some vocabulary and grammar for the analysis of multi-environment trials, as applied to the analysis of FPB and PPB trials. Euphytica 122, 477–490.
| Some vocabulary and grammar for the analysis of multi-environment trials, as applied to the analysis of FPB and PPB trials.Crossref | GoogleScholarGoogle Scholar |
van Eeuwijk FA, Boer M, Totir LR, Bink M, Wright D, Winkler CR, Podlich D, Boldman K, Baumgarten A, Smalley M, Arbelbide M, ter Braak CJF, Cooper M (2010) Mixed model approaches for the identification of QTLs within a maize hybrid breeding program. Theoretical and Applied Genetics 120, 429–440.
| Mixed model approaches for the identification of QTLs within a maize hybrid breeding program.Crossref | GoogleScholarGoogle Scholar | 19921142PubMed |
Vega CRC, Andrade FH, Sadras VO (2001) Reproductive partitioning and seed set efficiency in soybean, sunflower and maize. Field Crops Research 72, 163–175.
| Reproductive partitioning and seed set efficiency in soybean, sunflower and maize.Crossref | GoogleScholarGoogle Scholar |
Walsh B (2005) The struggle to exploit non-additive variation. Australian Journal of Agricultural Research 56, 873–881.
| The struggle to exploit non-additive variation.Crossref | GoogleScholarGoogle Scholar |
Walsh B (2014) Special issues on advances in quantitative genetics: introduction. Heredity 112, 1–3.
| Special issues on advances in quantitative genetics: introduction.Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DC%2BC2c3lsVGgsQ%3D%3D&md5=b12cff1fda2ec0a8985d136883f3f8e5CAS | 24326299PubMed |
Wang J, van Ginkel M, Podlich D, Ye G, Trethowan R, Pfeiffer W, DeLacy IH, Cooper M, Rajaram S (2003) Comparison of two breeding strategies by computer simulation. Crop Science 43, 1764–1773.
| Comparison of two breeding strategies by computer simulation.Crossref | GoogleScholarGoogle Scholar |
Weber VS, Melchinger AE, Magorokosho C, Makumbi D, Bänzinger M, Atlin GN (2012) Efficiency of managed-stress screening of elite maize hybrids under drought and low nitrogen for yield under rainfed conditions in Southern Africa. Crop Science 52, 1011–1020.
| Efficiency of managed-stress screening of elite maize hybrids under drought and low nitrogen for yield under rainfed conditions in Southern Africa.Crossref | GoogleScholarGoogle Scholar |
Williams ER, Matheson AC, Harwood CE (2002) ‘Experimental design and analysis for tree improvement.’ 2nd edn (CSIRO: Melbourne)
Williams ER, John JA, Whitaker D (2006) Construction of Resolvable Spatial Row-Column Designs. Biometrics 62, 103–108.
| Construction of Resolvable Spatial Row-Column Designs.Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DC%2BD287ltlWmtQ%3D%3D&md5=09698ef8d536091d4db00d20fd05afe8CAS | 16542235PubMed |
Wright S (1932) The roles of mutation, inbreeding, crossbreeding and selection in evolution. In ‘Proceedings of the 6th International Congress of Genetics’. Ithaca, New York. pp. 356–366. (Brooklyn Botanic Garden: Menasha, WI, USA)
Biography
Mark Cooper completed his BAgrSc and PhD degrees and a Graduate Certificate in Education at the University of Queensland. From 1990 to 2000 Mark taught courses in genetics and plant breeding at the University of Queensland and supervised graduate students pursuing MSc and PhD degrees in quantitative genetics, plant breeding and related topics. In 1997, Mark was recognized by the Australian Institute of Agricultural Science and Technology as the Young Professional in Agriculture. In 2000, Mark joined Pioneer Hi-Bred and now has technology development and leadership responsibilities as a DuPont Fellow. His research has focused on seeking working answers to questions of broad relevance to plant breeding, including: (1) What can we learn about the genetic architecture of quantitative traits? (2) How do plant breeding strategies achieve sustainable long-term genetic improvement for quantitative traits? (3) What technologies can improve the effectiveness of plant breeding methodology? These and other related questions are pursued to improve our understanding of the limits of predictability that can be achieved when applying plant breeding strategies to long-term sustainable improvement of agricultural systems. Results from these research efforts have been implemented and have resulted in the release of commercial maize products. His research efforts have been recognized with a number of industry awards, including two Pioneer Achievement in Research Awards and in 2013 the DuPont Bolton/Carothers Innovative Science Award.