

Synthesis of Highly Water-Soluble Adamantyl Phosphoinositide Derivatives*
Mark Gregory A , Meng-Xin Yin A , Malcolm J. McConville B , Eleanor Williams C , Alex N. Bullock C , Stuart J. Conway D , Antony W. Burgess E , Bruno Catimel E and Andrew B. Holmes A FA School of Chemistry, Bio21 Institute, University of Melbourne, 30 Flemington Road, Parkville, Vic. 3010, Australia.
B Department of Biochemistry and Molecular Biology, Bio21 Institute, The University of Melbourne, 30 Flemington Road, Parkville, Vic. 3010, Australia.
C Structural Genomics Consortium, University of Oxford, Old Road Campus Research Building, Roosevelt Drive, Oxford OX3 7DQ, UK.
D Department of Chemistry, Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford OX1 3TA, UK.
E Walter and Eliza Hall Institute of Medical Research, Parkville, Vic. 3052, Australia.
F Corresponding author. Email: aholmes@unimelb.edu.au
Australian Journal of Chemistry 68(4) 543-548 https://doi.org/10.1071/CH14543
Submitted: 2 September 2014 Accepted: 23 September 2014 Published: 3 December 2014
Abstract
Phosphatidylinositol phosphates are key regulators of cell signalling pathways and membrane trafficking in eukaryotic cells, and there is a need for new chemical probes to further understand how they interact with lipid-binding proteins. Here, the synthesis of phosphatidylinositol phosphate analogues containing adamantyl carboxylic ester groups, in place of the natural lipid side chains, is described. These derivatives are considerably more soluble in water than analogues containing other lipid side chains and do not form large aggregates such as liposomes or micelles. These adamantyl analogues bind to known phosphoinositide-binding proteins with similar affinities to native ligands and will facilitate future studies on the substrate specificities of these proteins involving cocrystallisation studies with proteins.
Introduction
Phosphatidylinositol phosphates (PtdInsPs or PIPs) play significant roles in multiple biological processes within the cell.[1–3] They function as precursors for secondary messengers in cell signalling pathways and act as membrane receptors for a variety of proteins involved in intracellular membrane trafficking and organelle biogenesis. These processes underlie normal cellular and tissue development, while defects can lead to disease.[4] The development of new probes for investigating phosphoinositide function is therefore of high importance from a biological and a pharmaceutical view point.[5–7]
There are eight known naturally occurring PIPs, varying only in the phosphorylation status of the 3-, 4-, and 5-positions of the myo-inositol head group of the phospholipid (summarised in Fig. 1). The polar head group of PIPs is linked to a diacylglycerol moiety via the myo-inositol C-1 hydroxyl through a phosphodiester linkage. The fatty acids found in naturally occurring PIPs have varying lengths and levels of unsaturation, and the precise constitution is species-dependent. In animals, the major molecular species contains stearic acid (18 : 0) at the sn-1 position and arachidonic acid (20 : 4) at the sn-2 position (as shown in Fig. 1). However, in plants, a range of carboxylic acids from palmitic acid (16 : 0) and stearic acid to linoleic acid (18 : 2) can be found at the sn-1 position with linoleic acid at the sn-2 position.[8,9]
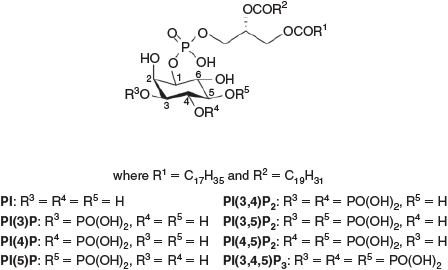
|
We have previously described the synthesis of all eight derivatives of the PIP family of compounds and analogues carrying an ω-N-terminal substituent on the sn-1 chain for each compound.[10] This modification was used to immobilised these lipids onto affinity beads for proteomic studies and onto a BiacoreTM chip for detailed evaluation of the affinity of different PIP-binding proteins for their cognate ligands.[11] The synthesis of these derivatives was made possible through the development of a reductive selective cleavage of myo-inositol orthoformate to allow regioselective protection of the six hydroxyl groups around the inositol ring.[12] Enantiomerically pure compounds were obtained through diastereomeric resolution, using camphanic derivatives, in order to achieve separation of the diastereomers.[8]
The synthetic derivatives of these molecules were used to form liposomes for characterisation purposes.[13,14] However, it is not always desirable to form liposomes or higher-order aggregates when studying such compounds. Furthermore, the long-acyl-chain derivatives are not particularly soluble in water owing to their hydrophobicity, which can cause problems when studying the interactions of such compounds with polar macromolecules. Derivatives with shorter alkyl chains such as butanoyl ester side chains have been developed and applied to protein cocrystallisation studies.[15] However, these compounds have also been reported to be unstable. Cocrystals of known PIP-binding proteins with the equivalent lipid-free inositol phosphate, such as myo-inositol 1,4,5-trisphosphate (IP3), have also been prepared but these analogues completely lack the diacylglycerol lipid moiety that is likely to contribute to the affinity of these compounds with certain proteins.
Here, the adamantane-carboxylic ester groups have been used as alternatives to the long fatty acid chains. Adamantane-carboxylic acid has been used previously to fabricate semisynthetic analogues of glycolipids.[16] The aqueous solubility of the resulting compounds has been reported to be significantly increased, which could be attributed to the formation of low-order aggregates.[16,17] In addition, the rigid globular hydrocarbon framework of adamantane groups may facilitate the crystallisation processes. Furthermore, they may also mimic the actual behaviour and conformation of the long acyl chains of naturally occurring phosphoinositides in vivo when exposed to the buffer-like cytosolic environments.
Results and Discussion
The adamantane-containing side chain phosphoramidite 5 was synthesised successfully from (+)-1,2-O-isopropylidene glycerol 1 (Scheme 1), which was also used as a precursor in the previous synthesis of the N-terminal, and unsaturated, side chain analogues of the full family of PIPs.[10] Diol 2 was obtained by paramethoxybenzyl (PMB) protection of 1 followed by subsequent methanolysis of the acetal group.[18] Exhaustive esterification of 2 with adamantyl chloride gave the desired PMB-protected lipid 3 in 86 % yield.
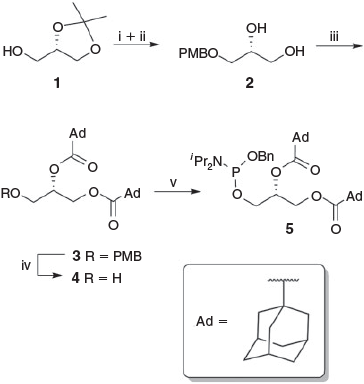
|
Traditionally, esterification of compound 2 with other acid chlorides forms the ester at room temperature but in this case, owing to the steric demand of the adamantane moieties, heating under reflux was required to induce the reaction to reach completion. Ceric ammonium nitrate (CAN) oxidation successfully removed the PMB group to afford the 1,2-di-O-adamantanecarbonyl-sn-glycerol 4 in 79 % yield. Phosphitylation of the resulting alcohol with the bisphosphoramidite (BnO)P(NiPr)2,[8] in the presence of 1H-tetrazole, provided the di-adamantyl glycerol-side chain phosphoramidite 5 in 90 % yield.
The protected inositol head group intermediates for both PI(4,5)P2 6 and PI(3)P 7 were synthesised in 13 and 10 steps respectively from myo-inositol.[10] Coupling the side chain phosphoramidite 5 with the C-1 hydroxyl derivatives 6 and 7 gave the fully protected intermediates 8 and 9 in moderate yields after purification by flash chromatography (Scheme 2). The final step employed global hydrogenolytic cleavage under pressure in the presence of a palladium catalyst.[8] In the case of the PI(4,5)P2 derivative, conditions using Pd black (20 equiv.) in a 6 : 1 tert-butanol/water mixture containing sodium bicarbonate gave the adamantane-PI(4,5)P2 derivative 10 as the sodium salt in 62 % yield. For the PI(3)P derivate, Pd(OH)2 was used in a 3 : 2 THF/methanol mixture to give the adamantine-PI(3)P derivative 11 in quantitative yield. Indicative 31P NMR signals for compound 11 at –0.35 and –0.90 ppm, shifted from further downfield, and no aromatic resonances in the 1H NMR spectrum confirmed that the benzyl protecting groups had been removed. A high-resolution mass spectroscopy (HRMS) peak of m/z 737.2336, corresponding to the (M – H)– ion (theoretical value 737.2336), confirmed the identity of the final product 11.

|
As predicted, these analogues were considerably more soluble in water than the naturally occurring compounds and no liposomes or micelle formation was observed even at high concentration. However, it was suggested that, owing to their bulk, the adamantyl groups could potentially affect the binding of the head group to the protein binding pocket. It was therefore important to test the binding nature of these compounds before further study.
Isothermal titration calorimetry (ITC) studies were carried out to compare the binding kinetics of the free head group (inositol 1,3-diphosphate) and the PI(3)P analogue 11. The affinity of compound 11 for the known PI(3)P-binding FVYE domain-containing protein HGS (hepatocyte growth factor-regulated tyrosine kinase substrate) shows only a small reduction in binding affinity when compared with the free head group (Fig. 2); dissociation constant (kD) values of 6.1 and 1.8 µM respectively were observed. Furthermore, this protein has a well-defined binding surface in comparison with other known PIP-binding domain-containing proteins, indicating that the adamantyl groups are indeed useful replacements of the naturally occurring long-chain fatty acids.[19]
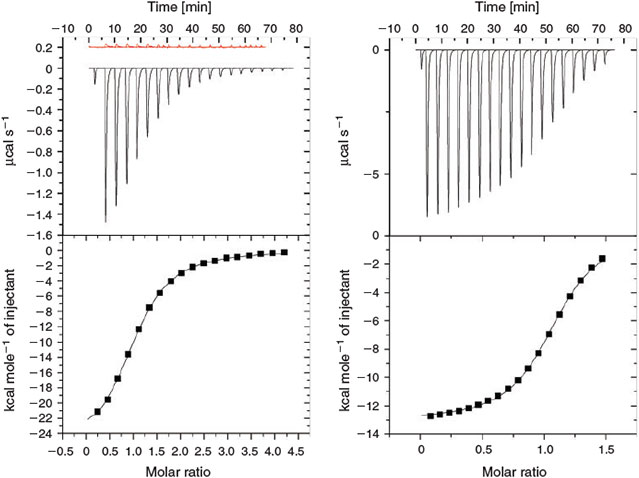
|
Conclusion
In conclusion, highly water-soluble derivatives of the naturally occurring phosphoinositides have been generated by replacing the acyl fatty chains with adamantyl groups. These analogues containing bulky adamantyl groups have been demonstrated to retain the binding affinity of the phosphorylated myo-inositol head groups for the FYVE domain-containing protein HGS. Studies are now focussed on obtaining cocrystallised structures in order to fully characterise the binding of these phosphoinositides with their protein counterparts and to determine the importance of the diacylglycerol unit in protein recognition.
Experimental
1H NMR spectra were recorded on Varian Unity 400 (400 MHz) or Varian Unity 500 (500 MHz) instruments, using deuterochloroform (or other indicated solvents) as reference or internal deuterium lock. The chemical shift data for each signal are given as δ in units of parts per million (ppm) relative to tetramethylsilane (TMS) where δ TMS = 0.00 ppm. The multiplicity of each signal is indicated by: s, singlet; br s, broad singlet; d, doublet; t, triplet; q, quartet; dd, doublet of doublets; ddd, doublet of doublet of doublets; dt, doublet of triplets; and m, multiplet. The number of protons (n) for a given resonance is indicated by nH. Coupling constants (J) are quoted in Hz and are recorded to the nearest 1 Hz. 13C NMR spectra were recorded on Varian Unity 400 (100 MHz) or Varian Unity 500 (125 MHz) instruments using the central resonance of the triplet of CDCl3 at δ 77.0 ppm as an internal reference. The chemical shift data for each signal are given as δ. 31P NMR spectra were recorded on Varian Unity 500 (202 MHz) instrument. The chemical shift data for each signal are given in units as δ ppm, using the chemical shift of 85 % H3PO4 at δ 0.0 ppm as the internal reference.
Infrared (IR) spectra were recorded on a Perkin Elmer Spectrum One Fourier-transform (FT)-IR spectrometer in the region 4000–650 cm–1. The samples were analysed as thin films from dichloromethane or as compressed solid samples.
Mass spectra were obtained on an LCMS system containing an Agilent 1100 HPLC and an Agilent 6220 electrospray ionisation time of flight (esiTOF) mass spectrometer fitted with a standard Agilent electrospray ion (ESI) source, in the Research Transfer Facility (RTF) at Bio 21 Institute, or a hybrid linear ion-trap and FT ion cyclotron resonance (FT-ICR) mass spectrometer (Finnigan LTQ-FT San Jose, CA), which is equipped with ESI, by the Mass Spectrometry Service at the University of Melbourne Chemistry Department.
Melting points were determined with an Electrothermal Engineering IA9100 or a Büchi 510 melting point apparatus and are uncorrected.
Analytical thin-layer chromatography (TLC) was completed on precoated 0.2-mm thick Merck Kieselgel 60 F254 silica gel plates and visualised by absorption of UV light and ethanolic phosphomolybdic acid (PMA) or aqueous potassium permanganate solution.
Flash chromatography was carried out, unless stated otherwise, on silica gel (Merck Kieselgel 60, 230–400 mesh) under a pressure of nitrogen.
Hydrogenations at high pressure were carried out in a Büchi GlasUster ‘miniclave drive’ stainless steel vessel, 100 mL, with a maximum operation pressure of 60 bar (6 MPa). Teflon inserts were used and reactions were stirred using magnetic stirrer bars.
Protein expression and purification involved human HGS protein (also known as Hrs, Uniprot O14964; residues 1–225), which was prepared from bacterial expression and purified by nickel-affinity and size-exclusion chromatography.
Isothermal titration calorimetry was performed at 15°C using a Microcal VP-ITC microcalorimeter. Proteins were buffered in 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.5, 100 mM NaCl. IP2 at 0.2 mM concentration was titrated into 0.02 mM HGS protein. Adamantyl lipid at 1.4 mM concentration was titrated into 0.2 mM HGS. Data were analysed using a single binding site model implemented in the Origin software package provided with the instrument.
Solvents such as n-hexane and N,N-dimethylformamide were purchased from Aldrich and dried over freshly activated 4-Å molecular sieves for 1 h. Anhydrous THF, diethyl ether, and dichloromethane were dried by passage through a packed column of activated neutral alumina under an argon atmosphere, and toluene by passage through a column with additional R3–11 copper-based catalyst (Pure Process Technology). Moisture levels of solvent from the drying columns were regularly checked by Karl Fischer titration. Petroleum spirits refers to the fraction of boiling point range 40–60°C. Procedures using moisture- or air-sensitive reagents were undertaken in a nitrogen-filled dual manifold employing standard Schlenk line techniques. Brine refers to a saturated aqueous solution of sodium chloride. Where appropriate and if not stated otherwise, all reactions were performed in flame-dried apparatus under an atmosphere of dry nitrogen.
3-O-Methoxybenzyl-1,2-di-O-adamantanecarbonyl-sn-glycerol (3)
To a stirred solution of diol 1 (500 mg, 2.13 mmol, 1 equiv.) in dry dichloromethane (10 mL) under nitrogen was added 4-dimethylaminopyridine (33 mg, 0.27 mmol, catalytic). The resulting solution was cooled to 0°C and dry pyridine (508 mg, 0.524 mL, 6.40 mmol, 3 equiv.) was added dropwise. After stirring for 30 min, adamantancarbonyl chloride (1.12 g, 5.88 mmol, 3 equiv.) was added dropwise. The reaction mixture was then warmed up to room temperature and heated at 65°C overnight. The reaction was quenched, once no starting material remained by TLC, by addition of water (30 mL) and the aqueous phase was extracted with dichloromethane (4 × 30 mL). The combined organic layers were washed with 2 M aqueous hydrochloric acid (30 mL) and the acid phase was back-extracted with dichloromethane (30 mL). The combined organic layers were washed with brine (35 mL), dried over MgSO4, filtered, and the solvent was removed under vacuum. Flash chromatography (10–20 % ethyl acetate/petroleum spirit v/v) afforded the diester 3 (983 mg, 1.83 mmol, 86 %) as a colourless solid.
Rf 0.25 (10 % ethyl acetate/petroleum spirit). 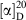 +7.38 (c 0.5, CHCl3). Mp 58–61°C. νmax (neat)/cm–1 2905, 2852, 1728, 1612, 1513, 1454, 1325, 1243, 1222, 1183, 1103, 1073, 1038, 819. δH (500 MHz, CDCl3) 7.24 (d, J 9, 2H), 6.86 (d, J 9, 2H), 5.23–5.17 (m, 1H), 4.49–4.44 (m, 2H), 4.33 (dd, J 12, 4, 1H), 4.14 (dd, J 12, 6, 1H), 3.80 (s, 3H), 3.55 (dd, J 5, 2, 2H), 2.03–1.96 (m, 6H), 1.86 (dd, J 19, 3, 12H), 1.74–1.66 (m, 12H). δc (125 MHz, CDCl3) 177.3, 176.9, 159.5, 130.1, 129.5, 114.0, 73.1, 70.0, 68.1, 62.7, 55.4, 40.9, 39.0, 39.0, 36.7, 36.7, 28.1, 28.1. HRMS (ESI+) m/z 559.3030; calc. for C33H44O6Na (M + Na)+ 559.3030.
+7.38 (c 0.5, CHCl3). Mp 58–61°C. νmax (neat)/cm–1 2905, 2852, 1728, 1612, 1513, 1454, 1325, 1243, 1222, 1183, 1103, 1073, 1038, 819. δH (500 MHz, CDCl3) 7.24 (d, J 9, 2H), 6.86 (d, J 9, 2H), 5.23–5.17 (m, 1H), 4.49–4.44 (m, 2H), 4.33 (dd, J 12, 4, 1H), 4.14 (dd, J 12, 6, 1H), 3.80 (s, 3H), 3.55 (dd, J 5, 2, 2H), 2.03–1.96 (m, 6H), 1.86 (dd, J 19, 3, 12H), 1.74–1.66 (m, 12H). δc (125 MHz, CDCl3) 177.3, 176.9, 159.5, 130.1, 129.5, 114.0, 73.1, 70.0, 68.1, 62.7, 55.4, 40.9, 39.0, 39.0, 36.7, 36.7, 28.1, 28.1. HRMS (ESI+) m/z 559.3030; calc. for C33H44O6Na (M + Na)+ 559.3030.
1,2-di-O-Adamantanecarbonyl-sn-glycerol (4)
To a stirred solution of the PMB-ether 2 (55 mg, 0.10 mmol, 1 equiv.) in acetonitrile (4 mL) and water (1 mL) was added CAN (337 mg, 0.61 mmol, 6 equiv.). After stirring for 2 h, the reaction mixture was diluted with water and ethyl acetate. The separated aqueous phase was extracted with ethyl acetate. The combined organic extract was washed with sat. NaHCO3 and brine, dried over MgSO4, and filtered. The solvent was removed under vacuum to give the crude product. Flash chromatography eluting with 10–20 % ethyl acetate/petroleum spirit afforded the alcohol 4 (33 mg, 0.079 mmol, 79 %) as a colourless film.
Rf 0.26 (20 % ethyl acetate/petroleum spirit). –1.82 (c 2.0, CHCl3). νmax (neat)/cm–1 2905, 2849, 1730, 1453, 1344, 1326, 1269, 1225, 1183, 1104 1072. δH (500 MHz, CHCl3) 5.07–5.03 (m, 1H), 4.30 (dd, J 12, 5, 1H), 4.20 (dd, J 12, 6, 1H), 3.71 (d, J 5, 2H), 2.04–1.98 (m, 6H), 1.89 (dd, J 9, 3, 12H), 1.76–1.66 (m, 12H). δc (125 MHz, CDCl3) 71.9, 61.8, 61.7, 40.8, 40.8, 38.8, 38.8, 36.4, 27.8. HRMS (ESI+) m/z 439.2455; calc. for C25H36O5Na (M + Na)+ 439.2455.
(+)-Benzyloxy(N,N-di-isopropylamino)(1,2-di-O-adamantanecarbonyl-sn-glycer-3-yl)phosphine (5)
A solution of benzyloxy-bis(N,N-di-isopropylamino)phosphine (548 mg, 1.62 mmol, 3 equiv.) and 1H-tetrazole (3.8 mL, 0.45 M, 1.73 mmol, 3.2 equiv.) in dry dichloromethane (5.0 mL) under nitrogen was stirred for 30 min. To the solution was added the diacyl glycerol 3 (225 mg, 0.54 mmol, 1 equiv.) in dry dichloromethane (25 mL). After stirring at room temperature overnight, the reaction mixture was diluted with dichloromethane (30 mL) and quenched by addition of sat. NaHCO3 solution (50 mL). The organic phase was separated and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The combined organic phases were washed with brine (50 mL), dried over MgSO4, filtered, and the solvent was removed under vacuum. Flash chromatography (5 : 15 : 80 triethylamine/ethyl acetate/petroleum spirit) afforded the phosphoramidite 5 (320 mg, 90 %) as a colourless oil.
Rf 0.9 (30 % ethyl acetate/petroleum spirit). 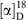 +5.7 (c 1.0, CHCl3). νmax (neat)/cm–1 2906, 2852, 1732, 1454, 1221, 1184, 1103, 1073, 1024, 976. δH (500 MHz, CHCl3) 7.42–7.30 (m, 5H), 5.18–5.12 (m, 1H), 4.76–4.59 (m, 2H), 4.35–4.30 (m, 1H), 4.16–4.10 (m, 1H), 3.82–3.60 (m, 4H), 2.00 (s, 6H), 1.88–1.86 (m, 12H), 1.73–1.66 (m, 12H), 1.22–1.15 (m, 12H). δC (125 MHz, CDCl3) 177.3, 176.9, 128.4, 127.4, 127.1, 70.8, 70.7, 65.6, 65.5, 65.4, 62.6, 62.5, 62.1, 61.9, 46.4, 43.3, 43.2, 40.9, 39.0, 36.7, 28.1, 24.9, 24.8, 24.7, 11.8. δP (202 MHz, CDCl3) 149.6, 149.5. HRMS (ESI+) m/z 676.3736; calc. for C38H56N1O6PNa 676.3738.
+5.7 (c 1.0, CHCl3). νmax (neat)/cm–1 2906, 2852, 1732, 1454, 1221, 1184, 1103, 1073, 1024, 976. δH (500 MHz, CHCl3) 7.42–7.30 (m, 5H), 5.18–5.12 (m, 1H), 4.76–4.59 (m, 2H), 4.35–4.30 (m, 1H), 4.16–4.10 (m, 1H), 3.82–3.60 (m, 4H), 2.00 (s, 6H), 1.88–1.86 (m, 12H), 1.73–1.66 (m, 12H), 1.22–1.15 (m, 12H). δC (125 MHz, CDCl3) 177.3, 176.9, 128.4, 127.4, 127.1, 70.8, 70.7, 65.6, 65.5, 65.4, 62.6, 62.5, 62.1, 61.9, 46.4, 43.3, 43.2, 40.9, 39.0, 36.7, 28.1, 24.9, 24.8, 24.7, 11.8. δP (202 MHz, CDCl3) 149.6, 149.5. HRMS (ESI+) m/z 676.3736; calc. for C38H56N1O6PNa 676.3738.
1D-2,3,6-Tri-O-benzyl-myo-inositol-(1′,2′-di-O-adamantanecarbonyl-glycer-3′-yl benzyl phosphate) 4,5-Bis(dibenzyl)phosphate (8)
1H-Tetrazole solution (0.29 mL, 0.45 M, 0.13 mmol, 3 equiv.) in acetonitrile was added to a solution of phosphoramidite 5 (63 mg, 0.095 mmol, 2.2 equiv.) in dry dichloromethane (1 mL) under nitrogen. After stirring at room temperature for 30 min, a solution of inositol head group 6 (42 mg, 0.043 mmol, 1 equiv.) in dry dichloromethane (1.2 mL) was added to the mixture dropwise. After stirring at room temperature overnight, meta-chloroperoxybenzoic acid (mCPBA) (140 mg, 70–75 %, 0.57 mmol, 6 equiv.) was added in a single portion and the mixture was stirred for a further 2 h. The reaction mixture was diluted with dichloromethane and washed with sat. aq. NaHSO3. The aqueous phase was extracted with dichloromethane and the combined dichloromethane extract was washed with sat. NaHCO3, brine, and dried (MgSO4). After filtration, the solvent was removed under vacuum to give the crude product. Flash chromatography (40–80 % ethyl acetate/petroleum spirit) afforded the protected lipid 6 (30 mg, 0.020 mmol, 47 %) as a colourless oil.
Rf 0.30 (50 % ethyl acetate/petroleum spirit). [α]D22 –3.6 (c 1.0, CHCl3). νmax (neat)/cm–1 2909, 2857, 1736, 1455, 1270, 1217, 1070, 1035, 1021, 737, 698. δH (500 MHz, CDCl3) 7.40–6.90 (m, 40H), 5.10–3.45 (m, 27H), 2.03–1.92 (m, 6H), 1.85–1.78 (m, 12H), 1.75–1.58 (m, 12H). δC (125 MHz, CDCl3) 176.7, 176.3, 138.2, 138.1, 137.4, 137.3, 136.1, 128.6, 128.5, 128.4, 128.3, 128.2, 128.1, 128.0, 127.8, 127.7, 127.6, 127.5, 127.2, 127.1, 127.0, 79.1, 79.0, 78.1, 78.0, 77.9, 77.7, 75.3, 75.2, 74.5, 74.4, 72.4, 72.3, 69.7, 69.6, 69.5, 69.4, 69.3, 69.1, 69.0, 68.9, 65.8, 65.5, 61.3, 60.3, 40.7, 38.7, 38.6, 36.4, 36.3, 27.8, 27.8, 27.7. δP (202 MHz, CDCl3) –0.6, –0.7, –0.9. HRMS (ESI+) m/z 1539.5914; calc. for C87H98O19P3 (M + H)+ 1539.5915; m/z 1561.5742; calc. for C87H97O19P3Na (M + Na)+ 1561.5735.
1D-2,4,5,6-Tetra-O-benzyl-myo-inositol-(1′,2′-di-O-adamantanecarbonyl-glycer-3′-yl benzyl phosphate) 3-(Dibenzyl)phosphate (9)
1H-Tetrazole solution (0.21 mL, 0.45 M, 0.93 mmol, 3 equiv.) in acetonitrile was added to a solution of phosphoramidite 5 (46 mg, 0.069 mmol, 2.2 equiv.) in dry dichloromethane (1 mL) under nitrogen. After stirring at room temperature for 30 min, a solution of 7 (25 mg, 0.031 mmol, 1 equiv.) in dry dichloromethane (2 mL) was added to the mixture dropwise. After stirring at room temperature overnight, mCPBA (100 mg, 70–75 %, 0.41 mmol) was added in a single portion and the mixture was stirred for a further 2 h. The reaction mixture was diluted with dichloromethane and washed with sat. aq. NaHSO3. The aqueous phase was extracted with dichloromethane and the combined dichloromethane extract was washed with sat. NaHCO3, brine, and dried (MgSO4). After filtration the solvent was removed under vacuum to give the crude product. Flash chromatography (25–75 % ethyl acetate/petroleum spirit) afforded the protected lipid 9 (19 mg, 0.014 mmol, 44 %) as a colourless oil.
Rf 0.24 (50 % ethyl acetate/petroleum spirits). [α]D –1.0 (c 0.6, CHCl3). νmax (neat)/cm–1 2908, 2854, 1730, 1455, 1269, 1218, 1071, 1017, 738, 696. δH (500 MHz, CDCl3) 7.65–7.17 (m, 35H), 5.14–4.70 (m, 13H), 4.54–4.48 (m, 1H), 4.37–3.99 (m, 7H), 3.94–3.89 (m, 1H), 3.77 (dd, J 9, 6, 1H), 3.57–3.45 (m, 2H), 2.04–1.85 (m, 6H), 1.83–1.77 (m, 12H), 1.75–1.58 (m, 12H). δC (125 MHz, CDCl3) 174.2, 173.8, 135.8, 135.5, 133.1, 126.0, 125.9, 125.8, 125.7, 125.3, 125.25, 125.2, 125.1, 124.9, 124.7, 124.6, 77.3, 75.3, 74.6, 74.3, 74.0, 73.5, 73.1, 72.8, 66.9, 58.9, 38.1, 36.2, 36.1, 33.9, 25.3, 25.2, 20.4. δP (202 MHz, CDCl3) –1.6, –1.67, –1.71, –1.75. HRMS (ESI+) m/z 1391.5586; calc. for C80H90O16P2Na (M + Na)+ 1391.5596.
1D-2,3,6-Trihydroxy-myo-inositol-(1′,2′-di-O-adamantanecarbonyl-glycer-3′-yl phosphate) 4,5-Bisphosphate (10)
To a solution of the protected lipid 8 (30 mg, 0.0195 mmol, 1 equiv.) in tert-butanol (4.2 mL) and water (0.7 mL) was added NaHCO3 (8.2 mg, 0.0975 mmol, 5 equiv.) followed by Pd black (41 mg, 20 equiv.). The reaction vessel (Büchi GlasUster hydrogenator) was degassed with N2 three times and then filled with hydrogen (150 psi [1 MPa]) and stirred for 65 h at room temperature. The mixture was centrifuged and the supernatant was removed. The residue was washed with ethyl acetate and centrifuged again to remove the starting material or by-products left. The solid residue was stirred with water (5 mL) and centrifuged and the aqueous supernatant was collected. This procedure was repeated three times (1300 g, 10 min). The combined aqueous phases were passed through a plug of Celite and freeze-dried to afford product 7 (11 mg, 62 %) as a fluffy colourless amorphous solid.
[α]D22 –7.0 (c 0.3, D2O). νmax (neat)/cm–1 3389, 2906, 1717, 1657, 1235, 1085, 979. δH (500 MHz, CDCl3) 5.35–5.27 (m, 1H, CH2CHCH2), 4.49 (dd, J 12, 3, 1H), 4.30–4.22 (m, 3H), 4.15–3.90 (m, 5H), 3.71 (dd, J 10, 3, 1H), 2.05–2.00 (m, 6H), 1.93–1.86 (m, 12H), 1.89–1.68 (m, 12H). δC (125 MHz, CDCl3) 180.2, 179.9, 77.8, 77.7, 75.9, 75.8, 71.3, 71.2, 70.9, 70.7, 70.6, 63.7, 62.6, 40.9, 40.8, 38.3, 35.8, 35.7, 27.5, 27.4. δP (202 MHz, CDCl3) 3.3, 2.0, –0.8. HRMS (ESI–) m/z 817.2026; calc. for C31H48O19P3 (M – H)– 817.2018; m/z 839.1841; calc. for C31H48 NaO19 P3 (M + Na – H – H)– 839.1828.
1D-2,4,5,6-Tetrahydroxy-myo-inositol-(1′,2′-di-O-adamantanecarbonyl-glycer-3′-yl phosphate) 3-Phosphate (11)
To a suspension of the protected derivative 9 (8 mg, 6 µmol, 1 equiv.) in methanol and THF (3 : 2) was added palladium(ii) hydroxide on carbon (20 wt-%, 8 mg), and the mixture was placed in a high-pressure hydrogenation vessel. The vessel was purged with nitrogen (10×, 3 bar [0.3 MPa]) and hydrogen (10×, 10 bar [1 MPa]) before stirring rapidly (900 rpm) at room temperature for 48 h. The reaction vessel was then purged with nitrogen (3×, 3 bar [0.3 MPa]) before filtration of the crude reaction mixture through Celite, rinsing with methanol (50 mL) and milliQ water (50 mL). All solvent was removed (under vacuum and by lyophilisation) and the crude product was suspended in diethyl ether (10 mL) and milliQ water (15 mL). The organic layer was extracted with milliQ water (3 × 10 mL) and the combined aqueous layers were lyophilised to give a pure colourless solid that was then stirred with sodium hydrogen carbonate (3 equiv.) in milliQ water overnight. The solution was then lyophilised to give the pure product 11 (4.0 mg, quantitative) as a fluffy colourless solid.
Mp 146–148°C (dec.) (lyophilised from water). [α]D –34.5 (c 0.42, D2O). νmax (neat)/cm–1 3363, 2906, 2851, 1727, 1452, 1224, 1035. δH (500 MHz, D2O) 5.45–5.35 (m, 1H), 4.64–4.52 (m, 2H), 4.37–4.27 (m, 1H), 4.26–4.01 (m, 4H), 3.96–3.82 (m, 2H), 3.54–3.43 (m, 1H), 2.18–2.05 (m, 6H), 2.05–1.90 (m, 12H), 1.90–1.74 (m, 12H). δC (125 MHz, D2O) 75.8, 75.6, 73.4, 71.2, 70.6, 70.5, 70.0, 63.9, 62.6, 61.4, 40.8, 38.3, 35.8, 27.5. δP (202 MHz, CDCl3) –0.35. –0.90. HRMS (ESI–) m/z 737.2336; calc. for C31H48O16P2 (M – H)– 737.2345.
Supplementary Material
HRMS and 1H, 13C, and 31P NMR spectra of all new compounds reported in this paper are available on the Journal’s website.
Acknowledgements
We thank the Australian Research Council for supporting this work through Discovery Project (DP1094497). We would also like to thank The University of Melbourne, Bio21 Institute, Agilent Technologies, and the Newton Abraham Trust (Oxford) for financial support. MJM is a National Health and Medical Research Council (NHMRC) Principal Research Fellow. The Structural Genomics Consortium (SGC) is a registered charity (no. 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, the Canada Foundation for Innovation, Genome Canada, GlaxoSmithKline, Janssen, Lilly Canada, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and the Wellcome Trust (grant no. 092809/Z/10/Z).
References
[1] G. D. Prestwich, Chem. Biol. 2004, 11, 619.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXktF2ru7k%3D&md5=47091aa5c5f77361d1da5bc3823af95cCAS | 15157873PubMed |
[2] B. V. L. Potter, D. Lampe, Angew. Chem. Int. Ed. Engl. 1995, 34, 1933.
| 1:CAS:528:DyaK2MXosFOqsrY%3D&md5=30cb8afb43aeac83a351e6ea82288465CAS |
[3] S. J. Conway, G. J. Miller, Nat. Prod. Rep. 2007, 24, 687.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtVGrurjO&md5=2701d5e23bff015247ddf010b62b47daCAS | 17653355PubMed |
[4] S. Ogino, P. Lochhead, E. Giovannucci, J. Meyerhardt, C. Fuchs, A. Chan, Oncogene 2014, 33, 2949.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXpvVylsL8%3D&md5=158dce416d811a79347a9cb6998ec69bCAS | 23792451PubMed |
[5] M. D. Best, H. Zhang, G. D. Prestwich, Nat. Prod. Rep. 2010, 27, 1403.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFyisLnO&md5=f66b1adec996f6717ec91fac7de7a620CAS | 20714465PubMed |
[6] C. P. Downes, A. Gray, J. M. Lucocq, Trends Cell Biol. 2005, 15, 259.
| 1:CAS:528:DC%2BD2MXjslKhsb8%3D&md5=0da751af7ffffa679c0d9886a57b72b1CAS | 15866030PubMed |
[7] T. Bunney, M. Katan, Nat. Rev. Cancer 2010, 10, 342.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXltV2ktb4%3D&md5=732d12975c72eb55604afc7bad3567deCAS | 20414202PubMed |
[8] M. Best, H. Zhang, G. Prestwich, Nat. Prod. Rep. 2010, 27, 1403.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFyisLnO&md5=f66b1adec996f6717ec91fac7de7a620CAS | 20714465PubMed |
[9] T. Balla, Physiol. Rev. 2013, 93, 1019.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhsVekurfJ&md5=2be4ddda13f6dda6e4f3b3d4f0f0be3aCAS | 23899561PubMed |
[10] S. J. Conway, J. Gardiner, S. J. A. Grove, M. K. Johns, Z.-Y. Lim, G. F. Painter, D. E. J. E. Robinson, C. Schieber, J. W. Thuring, L. S.-M. Wong, M.-X. Yin, A. W. Burgess, B. Catimel, P. T. Hawkins, N. T. Ktistakis, L. R. Stephens, A. B. Holmes, Org. Biomol. Chem. 2010, 8, 66.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFekurnI&md5=6124b49c3961c5b3a2000622bef3b8edCAS | 20024134PubMed |
[11] B. Catimel, E. Kapp, M.-X. Yin, M. Gregory, L. S.-M. Wong, M. Condron, N. Church, N. Kershaw, A. B. Holmes, A. W. Burgess, J. Proteomics 2013, 82, 35.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXmslekt7w%3D&md5=a6a8a28e1b97d874947057a15c9d674cCAS | 23416715PubMed |
[12] I. H. Gilbert, A. B. Holmes, M. J. Pestchanker, R. C. Young, Carbohydr. Res. 1992, 234, 117.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXotFaqsg%3D%3D&md5=c52e03183b3d277031faee45c3396d14CAS |
[13] B. Catimel, C. Schieber, M. Condron, H. Patsiouras, L. Connolly, J. Catimel, E. C. Nice, A. W. Burgess, A. B. Holmes, J. Proteome Res. 2008, 7, 5295.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlant7nI&md5=9b47ecaab5732d5ad9806aeaa03f8cbdCAS | 19367725PubMed |
[14] B. Catimel, M.-X. Yin, C. Schieber, M. Condron, H. Patsiouras, J. Catimel, D. E. J. E. Robinson, L. S.-M. Wong, E. C. Nice, A. B. Holmes, A. W. Burgess, J. Proteome Res. 2009, 8, 3712.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmvVKru7Y%3D&md5=5df33e1606950c01f376f05182f50284CAS | 19463016PubMed |
[15] M. J. Begley, G. S. Taylor, S.-A. Kim, D. M. Veine, J. E. Dixon, J. A. Stuckey, Mol. Cell 2003, 12, 1391.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXhsVOmuw%3D%3D&md5=b9450f849c59c9d9d31228e43d5d2c08CAS | 14690594PubMed |
[16] M. Mylvaganam, C. A. Lingwood, Biochem. Biophys. Res. Commun. 1999, 257, 391.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXit1yksLY%3D&md5=76decee722e1c779364ea665a6cc6d19CAS | 10198223PubMed |
[17] R. Mahfoud, M. Mylvaganam, C. A. Lingwood, J. Fantini, J. Lipid Res. 2002, 43, 1670.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XotFems7k%3D&md5=4dbbe6b9dab2b26237970bdffb9127d2CAS | 12364551PubMed |
[18] Z.-Y. Lim, J. W. Thuring, A. B. Holmes, M. Manifava, N. T. Ktistakis, J. Chem. Soc., Perkin Trans. 1 2002, 1067.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xis1GitbY%3D&md5=a3da23bef51c83822e4b34a0f076f8c3CAS |
[19] Y. Mao, A. Nickitenko, X. Duan, T. E. Lloyd, M. N. Wu, H. Bellen, F. A. Quiocho, Cell 2000, 100, 447.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXhsVOlur8%3D&md5=d1b064429f3be8433aca293d5cd5af61CAS | 10693761PubMed |
* Dedicated to the memory of the late Sir John (Kappa) Cornforth, who inspired everyone he worked with.