

The Biomimetic Inspiration for Renewable Hydrogen Fuel Production from Water Oxidation within Artificial Photosynthesis
Ron J. Pace A B and Rob Stranger AA Research School of Chemistry, College of Physical and Mathematical Sciences, Australian National University, Canberra, ACT 0200, Australia.
B Corresponding author. Email: Ron.Pace@anu.edu.au
Australian Journal of Chemistry 65(6) 597-607 https://doi.org/10.1071/CH11476
Submitted: 13 December 2011 Accepted: 3 February 2012 Published: 13 April 2012
Abstract
The thermodynamic constraints for the operation of the water oxidizing Mn4/Ca cluster within Photosystem II (PS II) are discussed. These are then examined in the light of the known redox chemistry of hydrated Mn-oxo systems and relevant model compounds. It is shown that the latest high resolution crystal structure of cyanobacterial PS II suggests an organization of the mono Ca tetranuclear Mn cluster that naturally accommodates the stringent requirements for successive redox potential constancy, with increasing total oxidation state, which the enzyme function imposes. This involves one region of the Mn4/Ca cluster being dominantly involved with substrate water binding, while a separate, single Mn is principally responsible for the redox accumulation function. Recent high level computational chemical investigations by the authors’ strongly support this, with a computed pattern of Mn oxidation states throughout the catalytic cycle being completely consistent with this interpretation. Strategies to design synthetic, biomimetic constructs utilizing this approach for efficient electrolytic generation of hydrogen fuel within artificial photosynthesis are briefly discussed.
Introduction
Artificial photosynthesis (AP) is a generic term, which embraces a range of novel technologies for non-polluting electricity generation, fuel production, and carbon sequestration, using solar energy. As the name implies, the inspiration is drawn from natural photosynthetic systems, which developed in organisms that were amongst the earliest known to exist on earth.[1] These natural systems are thus the product of an extremely long (>2.5 billion years) process of evolutionary refinement.
The aim of artificial photosynthesis is to technologically reproduce the components of natural photosynthesis on a large scale, for efficient solar energy conversion. The program offers the prospect of economic photovoltaic electricity generation and food production requiring negligible water usage compared with conventional agriculture.[2] However, in the near term, the most achievable goal is renewable, economically competitive electrolytic H2 generation from convenient water sources, such as seawater. This requires a suitable ‘super catalyst’ for the anodic, water oxidizing reaction, which is rate limiting in all conventional electrolytic systems generating H2. In fact nature has solved this particular problem, within photosynthesis, almost to the absolute limit of thermodynamic efficiency. Although the overall photosynthetic conversion of light into stored chemical energy (sugars, etc.) has many steps, with a total efficiency in nature of typically 4 % or less, the initial electron transfer steps, notably those related to water oxidation, are generally very efficient when viewed as isolated processes.[3]
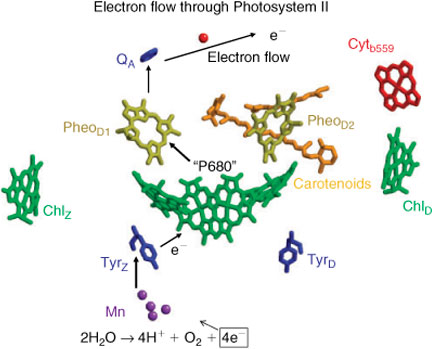
In all oxygenic photosynthetic organisms (plants, algae, cyanobacteria, etc.), the primary photosynthetic energy transducing processes generate an electron flux and an associated electrochemical gradient across the thylakoid membrane. The electrons come from water and this oxidation of water to molecular oxygen is the ultimate source of virtually all bioenergetic electrons utilized by living creatures. The process occurs within the oxygen evolving complex (OEC) located in Photosystem-II (PS II), a large multisubunit membrane-bound protein complex. The OEC comprizes an oxo-bridged Mn4/Ca cluster, with associated co-factors, notably a redox active tyrosine, Yz.[4] All oxygenic photosynthetic organisms share a common set of ‘core’ membrane proteins in PS II, which include the D1, D2 reaction centre (RC) proteins and inner chlorophyll a (Chla) binding proteins.[4] The D1/D2 heterodimer (analogous to the type II purple bacterial reaction centre[1]), contains the photo-oxidizable special chlorophyll pair, ‘P680’, as well as the pigment components associated with the primary light driven charge separation across the thylakoid membrane (Fig. 1). The D1 peptide principally ligates the catalytic Mn cluster responsible for water oxidation, near the donor (lumen in plants) membrane surface. Extra-membrane proteins, which are more organism specific, bind near the OEC and are believed to perform stabilization/regulatory functions.
|
Water Oxidation in PS II
The water oxidation process involves three distinct steps, each operating at an efficiency near the theoretical limit and unmatched by any related synthetic system (see Fig. 1):
-
Trapping of light energy by chlorophyll pigments and rapid energy transfer to the reaction centre (P680), resulting in its oxidation to P680+.
-
Rapid electron donation to P680+, through an oxidizable protein side-chain intermediate (tyrosine 161,YZ, on the D1 peptide), which stabilizes the charge separation in PS II.
-
Oxidation of water to molecular oxygen within the OEC. This is the most energetically demanding simple reaction performed by nature, requiring a redox potential per electron of ∼0.9 V (at pH ∼7). The redox potential developed in P680+ is in excess of 1.3 V; however, the YZ intermediate has an operating potential of only 1.0–1.1 V, barely above the thermodynamic limit for OEC operation.[5]
The OEC Mn4/Ca cluster is thus the most efficient anodic ‘electrolysis’ system known. It operates under mild conditions of temperature, pH, electrolyte background, etc., with a maximum turnover rate of ∼103 s–1 and effective overvoltage of at most ∼0.1 V.
It catalyzes the reaction:
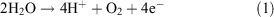
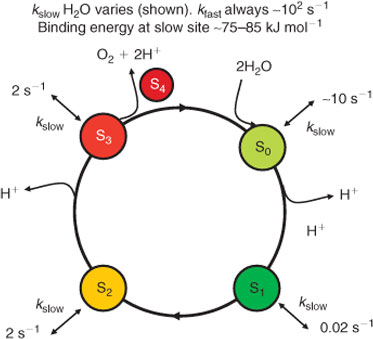
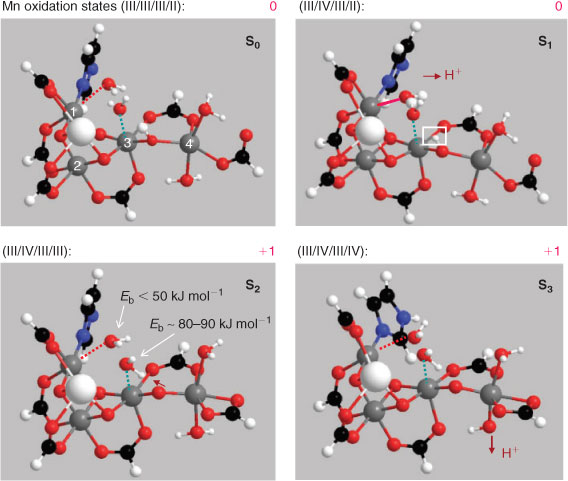
which proceeds through five intermediates consisting of four meta-stable states, labelled S0, S1, S2, and S3, and one short-lived ‘final’ state, S4, where the subscript refers to the number of stored oxidizing equivalents in the catalytic centre. Water remains exchangeable with this site up to S3, and the final oxidation of two water molecules to dioxygen occurs in a concerted, four electron step.[6] It is now generally held that the S states correspond to progressively increasing mean oxidation levels of the Mn ions, at least up to S3.[7] The sequence, with the accepted pattern of proton loss, is indicated in Fig. 2.
|
Much detail of the Mn4/Ca catalytic site has now been revealed by a recent crystal structure of cyanobacterial PS II to 1.9 Å resolution by Umena et al.[9] building upon earlier structures at lower resolution (2.9, 3.0, 3.5, and 3.7 Å).[10a–d] These structures should correspond to the ‘dark stable’ S1 state, although questions concerning radiation-induced Mn reduction during the X-ray diffraction (XRD) data collection process remain.[11]
The 1.9 Å structure allows a full experimental definition of the ligand environment of each metal in the OEC cluster, at least for a presumed formal S1 state. Although the protonation state of individually resolved O atoms is not revealed, the structure is closely consistent with recent computational models of the cluster,[12] derived from the protein derived ligation pattern revealed earlier by the 3.0 and 2.9 Å structures. Notable is the presence of several water/hydroxide/oxo molecules binding to the fourth (so called ‘dangler’) Mn, which has only two protein supplied (carboxylate) ligands. The necessity for such extra O moieties had been consistently inferred from computational modelling of the site by several groups.[13–15]
Fig. 3 shows a comparison of the immediate OEC region from the 1.9 Å structure and our most recent (Petrie et al. unpublished results) computational modelling of the Mn4/Ca cluster and ligands (truncated to ligating functional groups, as in ref. [13]). Mn numbering is according to our previous convention.[13] This model is a new, but close, variant of those we have described earlier,[13] which includes two hydroxides rather than waters near (or ligating to) the ‘dangler’ Mn(4), It is a member of a series we are examining to determine what effects such alterations may have on the detailed structure and distribution of oxidation states, within clusters maintaining the same mean Mn oxidation state for S1 (3.0). The model cluster is maximally ‘hydrated’, in that all potential direct water/hydroxide binding sites on the metals are saturated. In this instance the oxidation state distribution is Mn4III and this reproduces well the metal–metal distances seen in the 1.9 Å crystal structure. These distances are, however, generally longer in the two ‘short’ Mn–Mn vectors (2.8–2.9 Å) than those seen for the S1 state from extended X-ray absorption fine structure (EXAFS) measurements on PS II membrane samples and crystals with lower XRD resolution[9,10] (both vectors ∼2.7 Å). We have shown, however, that the latter geometry is also well reproduced computationally in clusters with the same mean Mn redox levels of 3.0, but with a III–IV–III–II oxidation pattern for Mn(1)…Mn(4). We believe this redox configuration to be that most consistent with a range of data on the photosystem, when functional PS II samples are conventionally prepared for study by spectroscopic and other techniques.[13c,13d]

|
It has long been realized, from X-ray absorption near edge spectroscopy (XANES) and photo-assembly measurements, that the Mn in the functional OEC site have oxidation levels significantly above MnII.[16] Furthermore, the S0 and S2 states have odd numbers of unpaired electrons in the exchange coupled cluster, with net spin 1/2 ground states arising from a predominantly antiferromagnetic coupling of the Mn ions. They exhibit Mn hyperfine structured signals in electron paramagnetic resonance (EPR) spectra at low temperatures (‘multilines’). This then dictates that the formal oxidation state in S1 is Mn4III or Mn2IIIMn2IV, or combinations equivalent to these. The mean oxidation levels in the other S states are then determined by adding or removing electrons. We have labelled the above two possibilities the ‘low and high’ oxidation state paradigms, respectively.[13d] At present, the high oxidation state assignment is generally favoured, based principally on empirical interpretation of Mn X-ray absorption spectroscopies applied to PS II in the S1 state and S states generated by single turnover flash advance.[7,16] However, we have recently shown, using a new time dependent density functional theory (TDDFT) approach, that the results from the most extensively used X-ray absorption technique, Mn K edge analysis, are consistent with the low oxidation state paradigm,[17] when metal ligand environment effects are computationally accounted for.
The fact that the available structural data on the Mn cluster geometry can also be accommodated within a low oxidation state model, particularly for the most studied S1 intermediate, is very significant when considering possibilities for the water splitting mechanism which operates within the site. To date all such detailed proposals, arising mainly from computational chemistry,[12,14,15] have assumed the high oxidation state paradigm and require one or both substrate water molecules to undergo progressive deprotonation throughout the S cycle. It is unclear, however, that this is consistent with the known pattern of substrate water exchange kinetics,[6] which we examine further below.
Water Oxidation and Mn Chemistry
Water oxidation occurs intimately within a local protein environment composed almost entirely of chemical species formed from hydrogens bonded to first row atoms (C, N, and O). These groups generally require similar energies for electron removal to that for water itself. The water splitting process may therefore be likened to ‘setting a fire in a wicker basket, without burning the basket’. There are two features of the OEC which then seem to us particularly relevant regarding its possible chemical mechanism.
-
Although the oxidized reaction centre, P680+, has a redox potential of ∼1.3–1.4 V, this is immediately ‘detuned’ to 1.0–1.1 V on passage through Yz, before communicating directly with the Mn cluster in the OEC, which must operate at a level close to 0.9 V.
-
The atoms coordinated to Mn and Ca in the OEC are almost exclusively oxygens, from hydroxy species, oxo bridges, and carboxylate protein side chains. There is only a single N-binding ligand (out of ∼21) from the His 332 imidazole side chain. This is the lowest known N/O ligation ratio for Mn in a protein, when the Mn oxidation states exceed II.
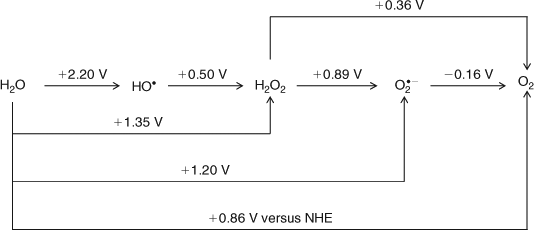
Examination of Scheme 1 (after Yamaguchi and Sawyer[18]) shows that the immediate ‘detuning’ of the redox potential by Yz serves to protect the ‘basket’, by essentially excluding the possibility of forming dangerous, reactive intermediate species like H2O2 or O2– (or even OH•), but at the cost of enforcing a concerted, four electron process for the water oxidation chemistry which must operate in an almost ‘activationless’ manner (i.e., low overvoltage). Furthermore, there is currently a large body of chemical experience with synthetic polynuclear Mn clusters of high (III, IV) formal redox level (e.g., see ref. [19]). Mostly, however, these have significant, often dominant levels of N ligation (N/O ∼2 per metal centre), along with oxo bridging. The N ligation helps to stabilize high Mn oxidation levels (IV, V), facilitating synthesis. By contrast, the predominantly O ligation in the OEC is notable because nature had the option to choose otherwise. Certainly N ligation to Mn, using histidine imidazoles or even heme or chlorin macrocycles (as in Jacobsen catalysts[20]) would have been easily possible in the photosystem protein matrix. Thus the high O ligation level is likely important, functionally. Because oxygen, either oxo or carboxylate derived, is a significantly harder ligand than N, so less stabilizing of high Mn oxidation states, the inference from this structural feature alone is that the Mn oxidation levels are likely to be at the lower, rather than higher, end of the above two ranges of oxidation state possibilities, while being consistent with the thermodynamic requirements for water oxidation.
|
Over the last decade, synthetic, self-assembled inorganic catalysts, based on Mn or Co, exhibiting efficient water electrolytic (or electro-photocatalytic) function have been developed.[21] Fig. 4A illustrates some examples. The catalysts have oxygen bridged polynuclear, extended structures, resembling in some cases known mineral forms. We have recently[22] surveyed these and noted the similarities within their repeating units of motifs strongly resembling the compact oxo-bridged core of the Mn ions in the OEC. We do not at present know which components of these structures actually bind water molecules or execute catalytic function. Nonetheless, some general reaction possibilities may be suggested from the known chemistry of Mn and Co, which we return to in more detail below.
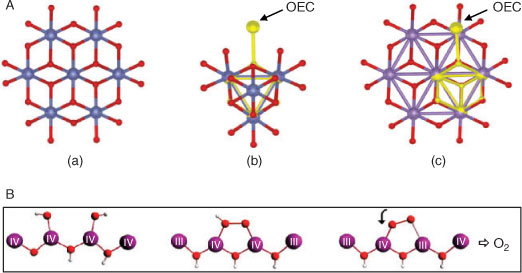
|
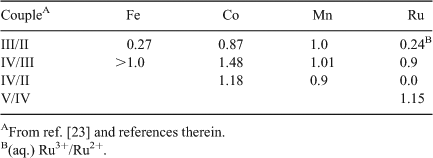
Prior to these developments, an extensive body of research had occurred (the previous two decades) on the use of deposited hydrated metal oxide surfaces for the anodic component of electrolytic cells (e.g., see ref. [23]). The most promising systems included oxides of the abundant transition metals from the first row, as well as Ru. Although detailed molecular and structural elucidation of these systems is yet to occur, important bulk physical characterizations were obtained: notably accurate mid-point potentials for successive redox changes of the metals in the hydrated oxide matrices. Some relevant data are given in Table 1. What is striking about these potentials is that, despite uncertainties of oxy-hydroxides being reasonable but not exact models for the OEC etc., only the Mn values are consistently poised, from oxidation level II to IV, to fit precisely into the narrow gap of redox potentials required for OEC function. To our knowledge, Mn is unique in the periodic table in this regard.
|
The basis of the ‘constant potential’ effect is presumably the ability of Mn centres in the hydrated oxide environment to compensate for charge increase by release of protons from probably terminally bound water/hydroxy groups, to maintain a near invariant redox potential of the ions with increasing formal oxidation state. This is consistent with the progressive oxidation behaviours of two broadly similar Mn oxo/alkoxo bridged binuclear complexes shown in Table 2. One possesses a terminal, deprotonatable H2O group ligated to a Mn, while the other does not.

|
Although the effect is not quite as dramatic as for the oxy-hydroxides, single proton loss from a terminal H2O group maintains successive Mn single electron redox potentials within ∼0.2 V, while the absence of such compensation causes the second single electron potential to be nearly a volt higher than the first. Also it is notable that the potentials for the L2 complex, which has a N/O ligand ratio of ∼0.6, are substantially larger, all else being equal, than those for the L1 complex, where the N/O ratio is 2. With predominant O ligation and the opportunity for charge compensation through proton loss from terminal hydroxy species, redox potentials are maintained close to 0.9 V, even for this (limited) system which is not particularly close, chemically, to the OEC Mn cluster. In total, the above observations speak closely of the existence of a broad, generic principle operating with oxo-Mn chemistry, which makes it uniquely suitable for operation within the water oxidase site in PS II. The Mn ligand environment should be mainly oxygen-based to establish potentials near 0.9 V for oxidation states from II to IV, and terminal H2O groups should (initially) be present to permit redox ‘levelling’ through progressive proton loss as the S states advance.
Examination of Table 1 shows that, although Mn is able to execute almost ‘exact’ redox levelling through proton loss, Co functions ‘well’ and this likely contributes to the effectiveness of the system discovered recently by Nocera et al.[21a] (Fig. 4A-a). In addition both Mn and Co share other features likely relevant to their utility in water oxidase catalysts. In their III and IV oxidation states, with predominantly O ligation, Mn and Co have redox potentials sufficient for four electron oxidation of water at neutral pH. XANES data show that the mean redox levels in the oxidized catalytic states are ∼+3.2 and ∼+3.7 for the Co phosphate and Mn birnessite catalysts, respectively.[25,21e] Redox potentials for Co in the II–III couple are likely similar to those for Mn in the III–IV couple (Table 1) within the layered hydrated oxides, so the two catalyst systems function as near equivalently poised. An additional important similarity then exists between the Mn and Co systems, whose detailed significance we are currently examining. Both ions have a co-ordinatively stable configuration of the 3d orbitals in a higher oxidation state (d3 for MnIV and low-spin d6 for CoIII), but a Jahn–Teller destabilized configuration in the next lowest oxidation level (high-spin d4 for MnIII and low-spin d7 for CoII). This allows a possible reaction pathway, illustrated schematically for the Mn case in Fig. 4B. An initial two-electron oxidation of two adjacently ligated water/hydroxy groups (the most difficult step) generates a peroxo group bound between two oxo-bridged metals in the higher oxidation state, with the electrons initially reducing other high-valent ions in the cluster. Internal electron (and possibly proton) transfer switches one of the peroxo bound metal ions to the lower, Jahn–Teller active oxidation state, which permits weakening of one metal–peroxo bond and then rapid final oxidation of the singly bound peroxo group to O2. For Co, this may involve facile interconversion of CoII between octahedral and tetrahedral coordination, a well known process in CoII chemistry.
Mechanistic Implications for the Mn Cluster in the OEC
Water molecules are thus expected to play two key roles in the catalytic function of the OEC. One obviously is to provide the substrate for the reaction, but the second, to provide the terminal ligand groups for redox levelling, is equally crucial. It is not obvious, however, whether these roles can be combined, the substrate species also providing the redox levelling through progressive proton loss, or whether distinct water molecules are separately employed for each function. Within the inorganic catalysts it seems reasonable that either could apply as there is no immediately obvious (to the authors) chemical advantage to either strategy. However, examination of the 1.9 Å OEC structure from Fig. 3 suggests that nature has chosen the second of the two above options. Four O groups, which are not bridges, are identified as being closest to the likely reactive portion of the cluster. Two (green box) are spatially close in a ‘cleft’ like region, involving Ca and Mn(1), Mn(3), and Mn(4), without being obviously fully ligated to any Mn. They are almost certainly waters, suggested by their bond lengths to nearby metals (2.4–2.8 Å). The second two (starred) are clearly ligated to Mn(4) alone, with bond lengths (∼2.1 Å) which indicate that they would probably be waters in the lowest S state(s). This arrangement is suggestive of ‘separate roles’ for the respective water species, with those on Mn(4) being the redox levellers. That there are two such groups on Mn(4) and that these are the only likely candidates for leveller function, from the XRD structure, means that Mn(4) might progress through two redox steps (i.e., from II to IV) as the S cycle advances. From the data in Table 1 this is plausible, while maintaining a cluster oxidation potential near 0.9 V.
The kinetics of substrate water exchange with the OEC site during S state turnover provide insight here. Hillier and Wyrzynski[6] have determined the exchange rates of substrate water with the OEC cluster in all S states. The results are summarized in Fig. 2. Basically, in all metastable S states, two distinct exchange rates are seen, a ‘fast’ and a ‘slow’ rate. The slow rate, which presumably corresponds to the more tightly bound substrate species and for which the data are best resolved, exhibits a counter-intuitive behaviour. It (and the fast rate) generally show no monotonic change in the exchange kinetics as might be expected from a progression of H2O, OH–, to O2– substrate species, bound to Mn sites of ever increasing oxidation level. Rather, the resolved kinetics are quite similar in all S states except S1, where the tighter bound water exchanges ∼100 fold more slowly than in the other states. These data appear to us particularly constraining and point strongly towards a mechanism in which the substrate waters are distinct from the redox levelling waters throughout the S cycle (to S3 at least).
We have shown computationally[13d] that this pattern of substrate water exchange is energetically consistent with the low oxidation state model and a cluster configuration in which the substrate waters are located as in Fig. 3, with neither directly ligating Mn(4), as is proposed in all other mechanistic models.[12] In S1 the strongly bound water actually becomes an hydroxide, through transfer of a proton to an oxo bridge joining Mn(3) and Mn(4), which reverses in S2 (see ref. [13d] and Fig. 5). Our published and most recent studies (summarized in Fig. 5) suggest that the two oxy species on Mn(4) are both waters in S0, but progressively deprotonate, so that by S3 at least one and probably both have become hydroxides, with one of these now forming an hydroxo bridge to Mn(3). This structure is consistent with EXAFS studies,[7,16] which detect a significant re-arrangement of the cluster in S3. The structural change has been interpreted as formation of an additional short (∼2.8 Å) Mn–Mn vector in S3, slightly longer than the two ∼2.7 Å vectors seen in S1 and S2.[16]
|
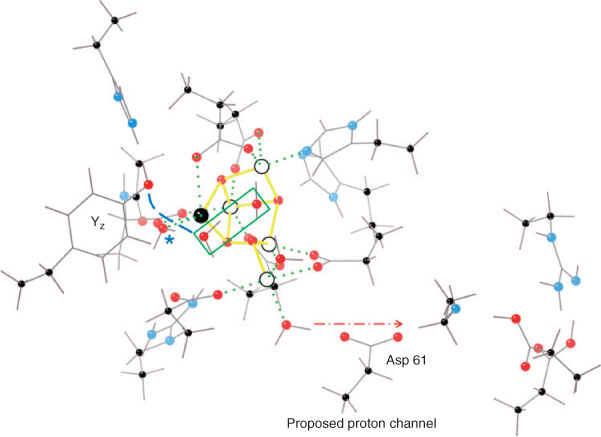
Fig. 5 also indicates the Mn oxidation state sequence suggested by our modelling. Consistent with the discussion above on redox levelling, it is Mn(4) that contributes most to redox accumulation up to S3, with Mn(1) and Mn(2) remaining invariant at the III level. Only Mn(2), which is ‘ligand saturated’ within the cluster and does not communicate directly with any exchangeable water/hydroxide group, undergoes oxidation (from the III to IV level) on the S0 → S1 transition. Thus, the general ‘insensitivity’ of the substrate water binding to the increasing level of redox accumulation in the OEC cluster is readily understood. Furthermore, proton loss in S3 is from the terminal water groups on Mn(4), adjacent to a likely proton exit channel commencing at Asp 61, which has been previously identified in the structure.[26] The putative proton channel also contains a Cl– ion. This proposal is further supported by computational modelling suggesting Asp 61 as the natural ‘proton exit’ point within the cluster (see ref. [27] and Fig. 6).
|
In S3 the cluster oxidation state configuration is III, IV, III, and IV. In principle, the transient S4 state could involve a further Mn oxidation, but evidence from several sources suggests now that this is unlikely (see ref. [12] for discussion). The ‘S4’ state is in fact a resolvable sequence, whose total duration of ∼50–200 μs, appears to be PS II preparation dependent,[28] involves several steps[29] including ∼1 proton release (t1/2 ∼ 30 μs)[30] and involves no detectable change in mean Mn oxidation level from S3 on the microsecond time scale.[7,30] Yz is very probably oxidized throughout, as there is no detectable kinetic distinction (at the tens of microseconds level) between O2 release and Yz• re-reduction.[31] Furthermore, at the beginning of the S4 ‘sequence’ the substrate is present as two water molecules, essentially as they existed in S3. This suggests that the actual, concerted four electron O2 forming reaction may be regarded formally as:
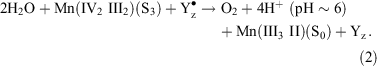
Based on the Mn hydrated-oxy data in Table 1 and the known potential for the Yz/Yz• couple, the ΔG for this reaction is ∼–0.6 eV. This is modestly exothermic, given the size of the free energy flows involved (∼4 eV), but entirely reasonable for a singularly demanding reaction which nature achieves with an efficiency we cannot yet match. Recent O2 back pressure measurements with PS II[32] indicate that the water splitting reaction is poised at least 200 mV above the reversible limit. At present there appears thus to be no thermodynamic need to invoke Mn oxidation levels higher than a mean value of 3.5 in S3 to achieve water oxidation.
Kinetically the O2 formation reaction proceeds very efficiently, with a low (probably <40 kJ mol–1) activation barrier. In particular, extensive computational studies by Siegbahn and others[14,15] have shown that the only facile mechanism for O–O bond formation satisfying the above reaction barrier limitation involves a formal O• radical attack on a water or hydroxy species, which our own work to date broadly supports. This step is readily achievable in the above cited, high Mn oxidation state models, because the substrate water(s) there undergo(es) progressive de-protonation during the S cycle (as proposed in these models), or enter effectively as hydroxide at a late stage. This contrasts with the picture advanced here, which is more naturally consistent with the OEC substrate water exchange behaviour, where these waters are fully protonated up to and including S3. However, managing the necessary proton removal–exchange during the S4 sequence, while retaining a sufficiently low reaction pathway energy profile, is then challenging.
We are currently engaged in a computational examination of possible reaction pathways for O–O bond formation, which are consistent with the above limitations. These will be reported separately, but already some factors are clear:
-
The calculations indicate that for O–O bond formation to occur, at least three protons must be removed from the substrate waters. The first is presumably lost (electrostatically) during the ∼30 μs step, following oxidation of Yz, before the main chemistry has commenced. Because of the progressive deprotonation of the terminal waters on Mn(4), one proton from the substrate waters may transfer, at minimal net energy change, to the leveller hydroxide on Mn(4). One further proton may readily transfer to the Mn(3)–O–Mn(4) bridge, a step which our modelling suggests actually occurs in S1, but then reverses (temporarily) in S2 and S3 (Fig. 5). The system obviously is ‘tuned’ to readily permit this transfer. It appears also necessary that one proton be transferred, probably in a coupled electron/proton step, to Yz•. The likely necessity for involvement of such a component in the water splitting reaction pathway has long been recognized.[33]
-
The resolved lag (up to ∼200 μs) in the step; S3 + Yz• → S0 + Yz appears to be the time required to establish the correct geometry for the coupled e–/H+ transfer to the phenoxy group of Yz•. Full DFT modelling of the local structure indicates that this must occur by proton exchange through one intervening water molecule (Fig. 6). These factors contribute to the larger re-organization energy required for electron transfer to Yz• on the S3 → S0 step, compared with the lower S state transitions, for which the Yz• re-reduction kinetics are generally an order of magnitude quicker.[31]
Towards the Design of Biomimetic Catalysts
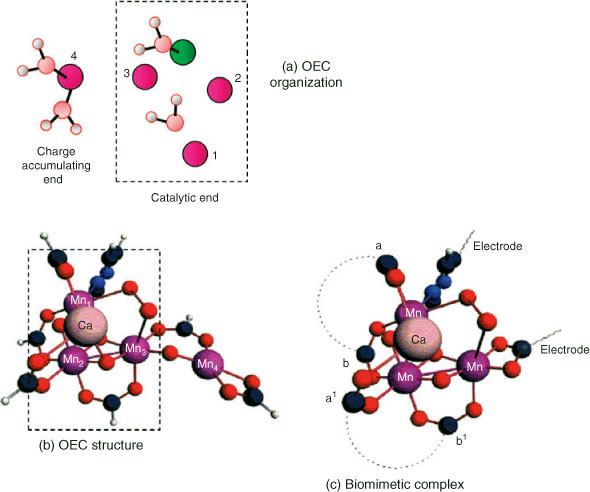
The ultimate aim of this work is to use our understanding of existing catalysts, particularly PS II, to design new catalytic components with activities approaching those of the natural system, but suitable for industrial-scale electrolytic H2 production. This will be challenging and directed by outcomes of our present investigations. The particular directions employed here into understanding the organization and catalytic detail of the OEC are unique and will drive our computational exploration of the possibilities. One novel strategy based on our understanding of the OEC to date is illustrated in Fig. 7. As outlined above, our computational work now strongly suggests that most redox accumulation actually occurs in Mn(4), outside the μ3-oxo-bridged sub-cluster (Fig. 7a). Thus Mn(4) acts as a ‘battery’ and therefore may be catalytically unnecessary. It might be replaced by another constant voltage electron sink (i.e., an electrode). A smaller metal cluster, such as that indicated schematically in Fig. 7c, could then function as the water oxidase. This system involves an open-edged cubane like our model of the OEC. Initially, computations will aim to establish:
|
-
The minimum nuclearity of the catalytic cluster, i.e., (Mn)3, (Mn)3/Ca, etc. A Mn dimer is not suitable,[34] but establishing function in a Mn trimer would have a huge synthetic advantage.
-
The nature and connectivity of the bidentate bridges. These could be phosphinate groups (like the cubane catalyst of Dismukes et al.[35]) attached to covalently linked aromatic or aliphatic moieties. Links could be chemically equivalent (ab, a′b′) or inequivalent (a′a, b′b) to enforce an open-edged structure.
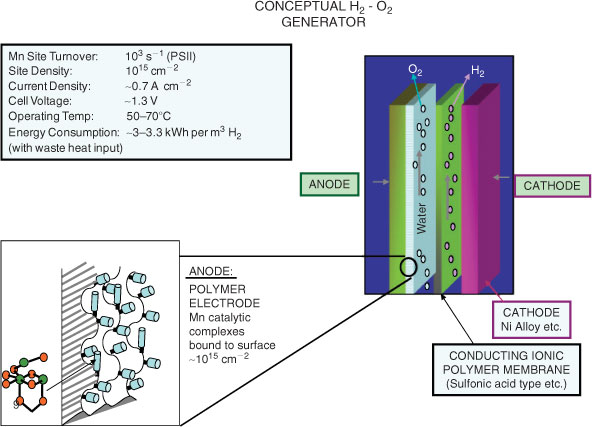
The system would operate at Mn redox levels equivalent to the OEC which is close to charge neutral. A functioning system could then be incorporated directly onto a conducting polymer/glassy carbon support, or some other immobilizing substrate surface (e.g, ref. [36], Fig. 8). This approach aims specifically to develop efficient catalysts for the anodic reaction in water splitting, which if successful might be useful in several applications requiring this type of electrochemistry. Thus Nocera et al.[37] have recently described an integrated system, involving both photo-electric components to generate electric current and catalytic surfaces (anode, cathode) for water electrolysis. The anodic surface is that derived from their in situ self-assembling and repairing Co phosphate surface. It seems likely to us, however, that any application involving catalysts of the type envisaged in Fig. 7 would be more suitable for large scale, high throughput systems, operating with electric power from any, including grid supplied, source (as in ref. [2]). Totally ‘light excluded’ operation should improve component lifetime, vital since flow through operation to limit gas mixing and promote convective proton flux through the two flowing liquid compartments would make a self assembling/repairing electrode surface, as in ref. [37], less straight forward to implement. In principle, if the immobilized biomimetic water oxidizing complexes operated at or near the efficacy of the OEC centre, in both turnover rate and over-potential, a high performance for H2 generation would be achievable. This is summarized in Fig. 8. The OEC actually operates in an environment with significant (×10–3 m) halide ion (Cl–) background, without anion oxidation. If this insensitivity to Cl2 generation could also be retained at high electrode current densities, the system in Fig. 8 could operate with a variety of readily available water sources.
|
Acknowledgements
R.S. and R.J.P. gratefully acknowledge financial assistance from the Australian Research Council. The authors also acknowledge the generous provision of supercomputing time on the platforms of the NCI (National Computational Infrastructure) Facility in Canberra, Australia, which is supported by the Australian Commonwealth Government.
References
[1] R. E. Blankenship, H. Hartman, Trends Biochem. Sci. 1998, 23, 94.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXisVOms74%3D&md5=fe6fc94aefa1b453e1d38d12b3767ee4CAS |
[2] R. J. Pace, An Integrated Artificial Photosynthesis Model, in Artificial Photosynthesis 2005, Chap. 2 (Eds A. Collings, C. Critchley) (Wiley-VCH: Weinheim).
[3] A. Melis, Plant Sci. 2009, 177, 272.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXpt1KgsrY%3D&md5=1e6610bab2a1457a8cfafa0d36e4ef8dCAS |
[4] K. Satoh, T. Wydrzynski, Govindjee, Photosystem II: The Light Driven Water: Plastoquinone Oxidoreductase 2005 (Springer: Dordrecht, The Netherlands).
[5] (a) (a) C. W. Hoganson, G. T. Babcock, Biological Processes, in Metal Ions in Biological Systems 1999, Volume 37, pp. 613–656 (Eds A. Sigel, H. Sigel) (Marcel Dekker: New York, NY).
(b) (b) D. A. Cherepanov, W. Drevenstedt, L. L. Krishtalik, A. Y. Mulkidjanian, W. Junge, in Photosynthesis: Mechanisms and Effects 1998, Volume II, pp. 1073–1076 (Ed. G. Garab) (Kluwer Academic Publishers: Dortrecht).
[6] W. Hillier, T. Wydrzynski, BBA-Bioenergetics 2001, 1503, 197.
| 1:CAS:528:DC%2BD3cXoslGjtb8%3D&md5=b6e7cef4fc14272af755ab99d0626d84CAS |
[7] M. Haumann, C. Müller, P. Liebisch, L. Iuzzolino, J. Dittmer, M. Grabolle, T. Neisius, W. Meyer-Klaucke, H. Dau, Biochemistry 2005, 44, 1894.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXmsVSksw%3D%3D&md5=838aa62173233b6ea9ac1f83035f2818CAS |
[8] F. Rappaport, J. Lavergne, Biochim. Biophys. Acta 2001, 1503, 246.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXoslGjtbo%3D&md5=8f4db7bbe82bde9d4406f8c08bde902eCAS |
[9] Y. Umena, K. Kawakami, J.-R. Shen, N. Kamiya, Nature 2011, 473, 55.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXkslCmtLg%3D&md5=f7b48594fae4a870a2b3d1c7ed10df43CAS |
[10] (a) A. Guskov, J. Kern, A. Gabdulkhakov, M. Broser, A. Zouni, W. Saenger, Nat. Struct. Mol. Biol. 2009, 16, 334.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhvFKjt7s%3D&md5=13b621fba48ee1ce230fbd2edab6997bCAS |
(b) B. Loll, J. Kern, W. Saenger, A. Zouni, J. Biesiadka, Nature 2005, 438, 1040.
| Crossref | GoogleScholarGoogle Scholar |
(c) K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber, S. Iwata, Science 2004, 303, 1831.
| Crossref | GoogleScholarGoogle Scholar |
(d) N. Kamiya, J. R. Shen, Proc. Natl. Acad. Sci. USA 2003, 100, 98.
| Crossref | GoogleScholarGoogle Scholar |
[11] S. Luber, I. Rivalta, Y. Umena, K. Kawakami, J.-R. Shen, N. Kamiya, G. W. Brudvig, V. S. Batista, Biochemistry 2011, 50, 6308.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXotVars7w%3D&md5=c265cdfd2761756bbb76a822fdda14f5CAS |
[12] P. Gatt, R. Stranger, R. J. Pace, J. Photochem. Photobiol. B 2011, 104, 80.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXntFWjtrg%3D&md5=ea42e23037a40d151517cd37512dfc19CAS |
[13] (a) S. Petrie, R. Stranger, R. J. Pace, Chem.–Eur. J. 2007, 13, 5082.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXntFyltr0%3D&md5=fa6f66631c30fa5d6abaf2295cfd1046CAS |
(b) S. Petrie, R. Stranger, R. J. Pace, Chem.–Eur. J. 2008, 14, 5482.
| Crossref | GoogleScholarGoogle Scholar |
(c) S. Petrie, R. Stranger, R. J. Pace, Angew. Chem. Int. Ed. 2010, 49, 4233.
| Crossref | GoogleScholarGoogle Scholar |
(d) S. Petrie, R. Stranger, R. J. Pace, Chem.–Eur. J. 2010, 16, 14026.
| Crossref | GoogleScholarGoogle Scholar |
[14] P. E. M. Siegbahn, Acc. Chem. Res. 2009, 42, 1871.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtlWmu7fJ&md5=0b1085c3dd673167528a43972b7804a2CAS |
[15] E. M. Sproviero, J. A. Gascón, J. P. McEvoy, G. W. Brudvig, V. S. Batista, J. Am. Chem. Soc. 2008, 130, 3428.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXitlCntL4%3D&md5=b4eda1587d6804886f333e1272a5b367CAS |
[16] V. K. Yachandra, K. Sauer, M. P. Klein, Chem. Rev. 1996, 96, 2927.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28Xmt1GlsL4%3D&md5=d03e8b8135d0835eebacf3f18c2f96bfCAS |
[17] (a) A. R. Jaszewski, R. Stranger, R. J. Pace, Phys. Chem. Chem. Phys. 2009, 11, 5634.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXotFWmsbc%3D&md5=1a930df933e5cf2ce9e99de37a2c2586CAS |
(b) A. R. Jaszewski, R. Stranger, R. J. Pace, Chem.–Eur. J. 2011, 17, 5699.
| Crossref | GoogleScholarGoogle Scholar |
[18] K. Yamaguchi, D. T. Sawyer, Inorg. Chem. 1985, 24, 971.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXhtFSjt7g%3D&md5=48e6508d1b98040a545ab8923e298dd6CAS |
[19] S. Mukhopadhyay, S. K. Mandal, S. Bhaduri, W. H. Armstrong, Chem. Rev. 2004, 104, 3981.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXnt12iur8%3D&md5=1a57249cc115931d6d3aeb8380925843CAS |
[20] E. Jacobsen, Acc. Chem. Res. 2000, 33, 421.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXjtFKhsLY%3D&md5=fb1833b0b2b90b6126cc62e4fcc113dfCAS |
[21] (a) M. Kanan, D. Nocera, Science 2008, 321, 1072.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtVSitrbP&md5=4d554995a8442318e37fd3fd4f64344fCAS |
(b) Q. Yin, J. M. Tan, C. Besson, Y. V. Geletti, D. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle, C. L. Hill, Science 2010, 328, 342.
| Crossref | GoogleScholarGoogle Scholar |
(c) F. Jiao, H. Frei, Angew. Chem. Int. Ed. 2009, 48, 1841.
| Crossref | GoogleScholarGoogle Scholar |
(d) D. M. Robinson, Y. B. Go, M. Greenblatt, G. C. Dismukes, J. Am. Chem. Soc. 2010, 132, 11467.
| Crossref | GoogleScholarGoogle Scholar |
(e) R. K. Hocking, R. Brimblecombe, L.-Y. Chang, A. Singh, M. H. Cheah, C. Glover, W. H. Casey, L. Spiccia, Nat. Chem. 2011, 3, 461.
[22] G. F. Swiegers, J. K. Clegg, R. Stranger, Chem. Sci. 2011, 2, 2254.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht12isrvL&md5=2bb497c792c79a23dde60032a1b3c096CAS |
[23] G. L. Elizarova, G. M. Zhidomirov, V. N. Parmon, Catal. Today 2000, 58, 71.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXisFyqt7k%3D&md5=5740faa55435e1b99b1ef741b07d70c3CAS |
[24] (a) T. Aderemi, R. Oki, J. Glerup, D. J. Hodgson, Inorg. Chem. 1990, 29, 2435.
| Crossref | GoogleScholarGoogle Scholar |
(b) M. T. Caudle, V. L. Pecoraro, J. Am. Chem. Soc. 1997, 119, 3415.
| Crossref | GoogleScholarGoogle Scholar |
[25] (a) M. W. Kanan, J. Yano, Y. Surendranath, M. Dincă, V. K. Yachandra, D. G. Nocera, J. Am. Chem. Soc. 2010, 132, 13692.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFGru7zM&md5=540cc424d400d53fa31ddfbaffecfcd3CAS |
(b) M. Risch, V. Khare, I. Zaharieva, L. Gerencser, P. Chernev, H. Dau, J. Am. Chem. Soc. 2009, 131, 6936.
| Crossref | GoogleScholarGoogle Scholar |
[26] (a) J. W. Murray, J. Barber, J. Struct. Biol. 2007, 159, 228.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXot1ygsrk%3D&md5=f9983b6703c87c7e46345cf254c4d179CAS |
(b) H. Ishikita, W. Saenger, B. Loll, J. Biesiadka, E.-W. Knapp, Biochemistry 2006, 45, 2063.
| Crossref | GoogleScholarGoogle Scholar |
(c) F. M. Ho, S. Styring, Biochim. Biophys. Acta 2008, 1777, 140.
[27] A. R. Jaszewski, R. Stranger, R. J. Pace, J. Phys. Chem. B 2011, 115, 4484.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjvVanu7g%3D&md5=fb61035972be193f134d93450ea6f7b3CAS |
[28] M. R. Razeghifard, R. J. Pace, Biochemistry 1999, 38, 1252.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXos12j&md5=7ebee8b998d8ec53e7b1364c891a5a7aCAS |
[29] J. Buchta, M. Grabolle, H. Dau, BBA-Bioenergetics 2007, 1767, 565.
| 1:CAS:528:DC%2BD2sXmt1ygtrY%3D&md5=33e704dc1b1ff60d130d32433c4762ddCAS |
[30] F. Rappaport, M. Blanchard-Desce, J. Lavergne, BBA-Bioenergetics 1994, 1184, 178.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXitFars78%3D&md5=902ac1e361aa55d891f82d5395e366e7CAS |
[31] (a) M. R. Razeghifard, R. J. Pace, Biochemistry 1999, 38, 1252.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXos12j&md5=7ebee8b998d8ec53e7b1364c891a5a7aCAS |
(b) M. R. Razeghifard, R. J. Pace, BBA-Bioenergetics 1997, 1322, 141.
| Crossref | GoogleScholarGoogle Scholar |
[32] D. Shevela, K. Beckmann, J. Clausen, W. Junge, J. Messinger, Proc. Natl. Acad. Sci. USA 2011, 108, 3602.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXivFKltL4%3D&md5=f96fad109a7f81c7d001a3e1b27091edCAS |
[33] C. W. Hoganson, N. Lydakis-Simantiris, X.-S. Tang, C. Tommos, K. Warncke, G. T. Babcock, B. A. Diner, J. McCracken, S. Styring, Photosynth. Res. 1995, 46, 177.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XnsFejsA%3D%3D&md5=23458410386145259fbdecf747cdc221CAS |
[34] C. D. Delfs, R. Stranger, Inorg. Chem. 2003, 42, 2495.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXitF2ru7w%3D&md5=01fcc169ab4dbcc491d1c78e313740d1CAS |
[35] G. C. Dismukes, R. Brimblecombe, G. A. N. Felton, R. S. Pryadun, J. E. Sheats, L. Spiccia, G. F. Swiegers, Acc. Chem. Res. 2009, 42, 1935.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVSgtb%2FL&md5=7ee1e63bbf20e924e0a4d165bd1f212dCAS |
[36] B. Cornell, G. Khrishna, P. Osman, R. Pace, L. Wieczorek, Biochem. Soc. Trans. 2001, 29, 613.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXms1SltLw%3D&md5=b7dc55aa2867b5d7345222206f58efddCAS |
[37] S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers, D. G. Nocera, Science 2011, 334, 645.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlyqu7vF&md5=e0298161b92e218b9b45c56538dc6eb5CAS |