

A Strategic, ‘Green’ Approach to Organic Chemistry with Microwave Assistance and Predictive Yield Optimization as Core, Enabling Technologies
Christopher R. Strauss A B CA QUILL Centre, The Queen’s University of Belfast, Northern Ireland BT9 5AG, UK.
B Strauss Consulting Ltd, PO Box 1065, Kunyung LPO, Mt Eliza, Vic. 3930, Australia.
C Corresponding author. Email: chris.strauss@qub.ac.uk

After 16 years investigating grape, wine, and spirit flavour until 1987, Chris Strauss pioneered fields that included microwave chemistry, green chemistry, predictive optimization of yields, and chemistry in high-temperature water. His group alone, created the market and the technological innovations that underpinned contemporary commercial microwave systems that now are employed extensively for chemical research and manufacturing globally. His prizes include the CSIRO Medal (shared), inaugural RACI Green Chemistry Challenge award and the Birch Medal. He presently shares his time among Queen’s University, Belfast, where he holds a Chair and conducts research, and in Europe and Australia, where he operates consultancies. |
Australian Journal of Chemistry 62(1) 3-15 https://doi.org/10.1071/CH08375
Submitted: 3 September 2008 Accepted: 22 October 2008 Published: 21 January 2009
Abstract
Since 1988, we have pursued enabling technologies and methods as tools for ‘green’ synthetic chemistry. The developed technologies comprise hardware including catalytic membranes and continuous and batch microwave reactors that have established global markets, as well as interactive, predictive software for optimization of yields and translation of conditions. New methods include ‘green’ reactions such as a catalytic symmetrical etherification, Pd-catalyzed coupling processes and a multi-component cascade for aniline derivatives. Reactions and workup were facilitated through solvent-free conditions, aqueous media at high temperature and dimethylammonium dimethylcarbamate (dimcarb) as a ‘distillable’ protic ionic liquid, as well as by non-extractive techniques for product isolation. The technologies and methods were designed for use alone or in various combinations as desired. Consolidation of individual operations or processes into unit steps was achieved through multi-tasking: media, reactants, catalysts, and conditions were selected to serve several purposes at various stages of a reaction. The tools were used to establish a technology platform comprising structurally diverse oligomers, macrocycles, and rod-like molecules supplementary to those available through phenol-formaldehyde chemistry. Dienone precursors were assembled from versatile building blocks containing complementary ‘male’ or ‘female’ fittings that were connected through inherently ‘green’ Claisen–Schmidt-type reactions. Isoaromatization afforded Horning-crowns, macrocyclic phenolic derivatives that were hybrids of calixarenes and crown ethers. Preliminary studies of organic substrates in salt water, with and without CO2, called into question proposals for disposal of anthropogenic CO2 by deep-sea dispersal.
Introduction
After 1900, chemical innovations afforded improvements in nutrition, agriculture, health, oils, materials, composites, and coatings.[1] Substantial increases in life expectancy and living standards ensued.[2] The chemicals industry earned strong public acceptance and support as a result. Those circumstances changed after 1962, however, when Rachel Carson in Silent Spring linked the indiscriminate use of agricultural pesticides and herbicides to adverse ecological events, predominantly affecting birdlife.[3] She also associated aerial spraying of crops with mass kills of fish. In the USA, a major fish kill in Mississippi in 1964, a chemicals fire on the Cuyahoga River, Ohio, in 1969, coupled with improper disposal of chemical residues, e.g. at Love Canal, New York, and at Times Beach, Missouri, broadened Carson’s message.[4] Disastrous explosions in chemical plants in Seveso (Italy), Flixborough (UK), and Mexico City in the 1970s were followed by the worst industrial accident in history in Bhopal (India) in 1984. The nocturnal escape of methyl isocyanate from a chemical plant left up to 10000 neighbouring residents dead and 50000 permanently injured.[5]
Negative sentiment toward the chemicals industry gained momentum in the 1970s and 1980s. Public opinion polls in Britain revealed a widely held perception that manufacturers were indifferent toward safety and the environment. Student enrolment in chemical courses decreased.[4] An executive of a chemicals corporation coined the word chemophobia to mean ‘the almost spontaneous negative response that occurs when people hear the words chemicals and chemical company’.[6]
The Bhopal disaster triggered a revamping of priorities in the industry, with safety and the environment assuming the highest.[4] Twenty years ago, however, synthetic chemists were ill-prepared and poorly equipped to meet these rapidly emerging challenges. Synthetic preparations of fine chemicals and pharmaceuticals typically generated in the order of 25–100 times more waste than product.[7] Equipment consisted mainly of glassware and ancillary apparatus the likes of which had been deployed for half a century or more. Protection and deprotection as well as hazardous or toxic solvents and reagents were commonly used. Temperatures well below 0°C tended to be those of choice and usually were obtained by wasteful methods.
With pharmaceutical companies concentrating primarily on ‘greener’ routes to their own products, we began to pursue the establishment of broadly applicable synthetic tools for cleaner and more efficient preparations.[8] The entire synthetic cycle lay within our ambit, including selection of starting materials, methods for performing reactions (the reaction, medium, catalyst, apparatus, conditions, and workup), the products and their properties, and avoidance or recycling of waste.
In the 1990s, progress was reported in invited reviews concerning the establishment of microwave chemistry[9] and environmentally benign chemistry,[8] a field now known as green chemistry.[10] The present review constitutes the final instalment in the series and, as with its predecessors, the emphasis is mainly on our activities.
Beginnings of Microwave-Assisted Organic Chemistry
The first disclosures of microwave-assisted organic synthesis came from Gedye, Giguere, and their respective coworkers in 1986.[11,12] Up until then, technologies for practical organic synthesis had seen little change in decades. Rate enhancements of up to three orders of magnitude afforded corresponding savings in time. Unfortunately though, these attractive results were offset by serious hazards, including explosions. Both groups established protocols for trying to manage the technique more safely,[11–13] but importantly, neither attempted to control or to further develop it.
Others interpreted the results to suggest that microwave heating of organic solvents was dangerous. They explored solvent-free microwave reactions, particularly with ‘dry’ media in open vessels.[14–31] Organic reactants, often together with an inorganic reagent, were adsorbed onto solid supports and heated in domestic microwave ovens. Samples were not mixed and their temperature was not measured. Results were not always reproducible between laboratories. As with the work of Gedye and Giguere, the methodology enabled management of reactions but not control. It was widely explored in the 1990s and reviewed extensively.[32–36]
Our efforts were not directed toward avoiding or managing the problems, but to overcoming them. The aim was to conduct organic processes safely and controllably in organic solvents if desired, under measurable, reproducible conditions, including at pressures above ambient. Dedicated, closed-vessel microwave reactors required to meet such objectives were designed and built.[37–39] For the decade up to 1998, the research domain remained ours exclusively. Proponents of solvent-free methods derided the approach, some going so far as to predict that microwave systems operating with volatile organic solvents would never be acceptable.
A prototype continuous microwave reactor (CMR) for flowthrough reactions was invented in 1988.[37] It satisfied our key safety and operating criteria. By 1995, a complementary laboratory-scale microwave batch reactor (MBR) with pressure-resistant vessels had been constructed.[39] It superseded our 1992 prototype[38] and enabled rapid heating (typically 1–2°C per second) and infinitely variable control of microwave power, as well as measurement of absorbed and reflected microwave energy. A load-matching device (cavity tuner) optimized heating efficiency. Temperature and pressure were measured directly by optic fibre thermometry and through a transducer, respectively. Magnetic stirring of the sample, unprecedented for microwave chemistry before our work,[38,39] ensured uniform temperature by thorough mixing. Chemicals could be added and samples withdrawn while heating. All wettable surfaces and connections were chemically inert. Other facilities included plumbing for gases and a cold finger for rapid, post-reaction cooling. Safety devices and protocols extended from materials of construction, the design of flanges, disks and seals, the microwave cavity, vessels and their containment, to electronic interlocks and emergency shut-down. Microwave power input was computer-controlled. Heating could be carried out at high or low rates as required and designated temperatures could be maintained for hours.
These units facilitated the introduction of concurrent heating and cooling (now often referred to by others as simultaneous heating and cooling) and differential heating for microwave chemistry.[9] Differential heating enabled immiscible phases composed of good and poor microwave absorbers to be heated to substantially different temperatures simultaneously in the one vessel, which may have been a flowthrough tube if desired. It was ideal for processes in which the starting material reacted in the hotter phase to afford a thermally labile product that could be extracted immediately into the cooler phase.[9,40]
Synthetic organic chemists consider that reaction time is approximately halved for each 10°C increase in temperature. This approximation, when applied to a process requiring 18 h at 80°C indicates that only 16 s would be required at 200°C (i.e. 212 times faster) provided that the components survived the higher temperature. The MBR and CMR conveniently exploited such efficiencies in time and energy, realizable by safely raising the temperature ~100°C above that attainable in the same solvents under reflux conditions. Reflux processes were transformed into continuous operations or rapid batch microwave reactions according to requirements. At pressures of 2–3 MPa, temperatures in the order of 200°C were obtained in the CMR and MBR with MeOH, EtOH, EtOAc, CHCl3, MeCN, and Me2CO for example, all of which boil below 85°C at atmospheric pressure. After opening the vessel, reaction products could be concentrated by rotary evaporation and the solvent redistilled for re-use.
Resistance-heated autoclaves also may be employed for such procedures, but not as readily. They are usually constructed with thick-walled metal vessels, heated externally. Conductive and radiative heat losses increase with temperature. Insulation can lessen the problem but would not be expected to curtail it. Owing to high thermal inertia of the vessels, heating to high temperatures and subsequent cooling usually require more time than with microwave systems where such operations typically could be completed within a few minutes.[9] Temperature gradients are difficult to avoid with resistance-heated systems. Pyrolytic degradation of components on or near the inner walls is common.
Vessels for the CMR and MBR were made from microwave-transparent insulating materials. Direct, bulk heating, combined with efficient mechanical stirring or mixing of the sample, minimized temperature gradients. The energy was absorbed directly by the reaction mixture in preference to the container. Thus, the vessel usually was no hotter than its contents, minimizing pyrolytic wall effects. Microwave power also could be applied or withdrawn instantaneously and the input could be adjusted readily to match that required.[8,9]
Advantages of the CMR and MBR included:
-
Thermally unstable products formed under microwave conditions could be rapidly cooled and isolated.
-
Samples could be withdrawn for analysis while material was being processed. With the CMR, reaction mixtures could be subjected to multiple passes if required, or the conditions altered instantaneously during a run.
-
Laboratory-scale batch reactions often could be readily conducted under flowthrough conditions in the CMR.
-
Moderate to high temperature reactions could be carried out in vessels fabricated from inert materials such as perfluoroacetoxy Teflon, polytetrafluoroethylene (PTFE), or quartz.
-
In multi-phase systems, selective heating was common.
-
Reaction times could be dramatically shorter than with conventional heating.
-
Energy was introduced remotely, without physical contact between the source and the sample.
-
Energy input to the sample started and stopped at the press of a switch.
-
Thermal inertia is lower than with conventional conductive heating.
-
Energy was delivered throughout the product, not predominantly at surfaces.
-
Heating rates were higher than could be achieved conventionally if at least one of the components could couple with microwaves.
Commercialization of Microwave Reactors
By 1995, we had performed hundreds of reactions safely by MBR and CMR.[9,37,39] Examples included acetalization, amidation, decarboxylation, esterification, etherification, hydrolysis of esters and amides, isomerization, oxidation, and transesterification. HOAc, Me2CO, MeCN, MeCOEt, PhCl, CHCl3, Me2NCHO, DMSO, EtOH, EtOAc, i-PrOH, MeOH, pyridine, THF, and PhMe contributed to a long list of solvents employed. Volatile reactants or gases such as CO2, Me2NH, and HCHO were processed without difficulty, including in cases where the vessel was pressurized with gas before heating. Over-pressure triggering emergency shut-down rarely occurred, even when gases were used as starting materials or when they were formed during reactions.
Procedures previously requiring hours or days at reflux were conducted reproducibly, under defined conditions within minutes. A typical example involved isopropyl mesitylate, a sterically constrained ester that was produced in 56% yield after 1 h at 148°C in the MBR. Conventional heating in refluxing i-PrOH afforded only 2% conversion after 28 h.
Microwave-transparent reaction vessels, made from chemically inert, insulating polymeric materials like PTFE, had inherent advantages for cleaner processing.[8] PTFE resists attack by strong bases and, unlike stainless steel, is not corroded by halide ions. Low adhesivity helped to minimize detergent and organic solvent usage during cleaning operations. Short reaction times also helped to reduce waste. Coolants usually were used only after the reaction, in low volume, and could be recycled.
These prized advantages and capabilities were unique at the time.[8,9,37,39] Accordingly, a sizeable market for pressure-resistant microwave reactors was created.[41] By 2000, after technology transfer, systems embodying the principles, concepts and designs of the CMR and MBR were released commercially. Manufactured in Europe, Asia, and in the USA, they now are distributed globally by several manufacturers and suppliers.[41–44] Sales for 2003 were estimated independently at ~$US 89 million.[42]
Scales for commercial microwave reactors range from <1 mL to ~2 L for batch systems, which can incorporate parallel syntheses in multiple vessels within a carousel.[42,43] Volumes up to 10 mL are typical for new compound discovery, where banks of automated reactors are employed. CMRs have throughputs up to ~100 mL per min. Typically, upper temperature limits lie between 180 and 230°C depending on the specifications for materials of construction of the vessels and fittings. Quartz vessels can accommodate higher temperatures. Examples of commercial systems based on the MBR and CMR are depicted in Fig. 1. Specialized systems for combinatorial chemistry, parallel synthesis (including multi-well titre plates), peptide synthesis, and for low-temperature reactions have been introduced.[44]

|
A New Paradigm for Synthetic Organic Chemistry
Of ~2 million organic synthetic reactions reported up to the year 2000, ~1.6 million had been carried out between 0 and 110°C. Times for >50% of examples were 1, 2, 3, or 4 h.[41] These data indicate that temperatures between 100 and 300°C and reaction times below 30 min had been employed relatively infrequently over the preceding century and a half. This circumstance appears to be largely attributable to the operational limits of the vessels and ancillary equipment that were state-of-the-art during that period.
Now, pressure-resistant closed vessel microwave reactors are used routinely for rapid reactions at significantly higher temperatures than 110°C. A 100-fold reduction in reaction time enables a 5-h process to be completed in only 3 min. Decreases of 1000-fold in reaction times previously required for traditional processes are common. Additional efficiencies are realized through process intensification. More reactions can be completed within a given time in fewer vessels than with traditional equipment. Automation has extended the synthetic chemist’s working day to 24 h if desired. Often yields are improved and product mixtures are less complex than with slower processes. Other significant benefits include energy savings, use of less or no catalyst, low rates of decomposition and enhanced opportunities for recycling solvents or media. Collectively, these developments and others discussed below indicate a paradigm shift in contemporary approaches to organic synthesis.
Reactions Requiring High Temperature
Closed vessel microwave reactors delivered improved conditions for established reactions known to require high temperatures. Willgerodt reactions, for example, involve heating of alkyl aryl ketones with sulfur and aqueous ammonia to form carboxylic acid amides having the same number of carbon atoms. Traditionally, they were conducted over several hours in an autoclave or bomb at ~200°C. With the MBR, they were completed in ~10 min at similar temperature and in comparable yields.[45]
The microwave reactors also enabled discovery of new reactions that require high temperature. First reported in 1852, Williamson etherification requires strongly basic conditions and generates an equivalent of salt as waste. Although not a green process, for wont of a better alternative, it remains one of the most significant and longest-surviving stoichiometric reactions.
A symmetrical etherification usually carried out at ~200°C was developed (Scheme 1).[46] It employed excess alcohol (ROH) and the corresponding alkyl halide (RX) as a catalyst that was repeatedly sacrificed and regenerated in situ. A solvolytic displacement reaction between RX and ROH afforded R2O and HX (Eqn 1). The liberated HX reacted with another molecule of ROH to form water and RX (Eqn 2). If reactions in Eqns 1 and 2 proceeded at comparable rates, the overall process (Eqn 3) utilized ostensibly neutral conditions, required participation by X– (which was present in catalytic quantities after the reaction started) and involved condensation of two molecules of ROH to give R2O plus water.
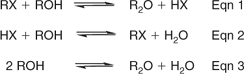
|
For Eqn 1 in Scheme 1, the alcohol acted as a solvent, reactant and nucleophile and for Eqn 2, as a solvent, electrophile and base. The halide needed to be an effective leaving group in Eqn 1 as well as a powerful nucleophile and weak base to satisfy Eqn 2. Although lacking the generality of Williamson etherification, advantageously, the symmetrical process was catalytic and did not produce salt.
Kinetic Products
Some reactions have the potential for generating both kinetic and thermodynamic products. Kinetic products are formed early in reaction pathways and they may not be stable. Their formation may be readily reversible or they may undergo further reaction to afford other kinetic or thermodynamically more stable products. Thermodynamic products may, but do not necessarily arise through reaction of kinetic intermediates. They can result from the starting material directly, by a competing, but slower process.[47]
Being typically transient, kinetic intermediates and products often are difficult to prepare efficiently. Heating rate is influential if reactions can proceed to more than one product by discrete pathways, especially if the reaction that proceeds first (i.e. that with the lower activation energy) is undesired. In such circumstances, rapid microwave heating can be advantageous, as demonstrated by Stuerga et al. for the sulfonation of naphthalene to give regioisomers of naphthalenesulfonic acids.[48,49] Rapid heating to 130°C afforded the 2-naphthalenesulfonic acid almost exclusively, whereas low heating rates gave a mixture of both isomers. Bose et al. controlled the steric course of β-lactam formation by varying the microwave power input. In the example illustrated in Scheme 2, after heating for 1 min, the trans to cis isomeric ratio was 1:5, but after 4 min, the cis isomer predominated by 6:5.[50]
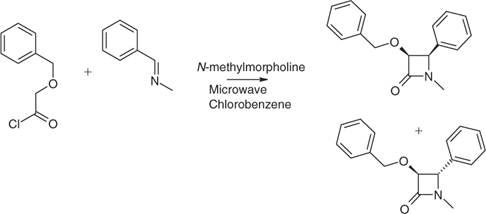
|
Instruments capable of rapid heating and cooling of reactions provide scope for such applications. Examples from our laboratories included the hydrolysis of cellulose to glucose in respectable yield[9] and studies into the chemistry of carvone,[51] allylphenyl ether,[52] and hexane-2,5-dione[53] in high-temperature water.
Fischer–Helferich glycosylation involves acid-catalyzed condensation of a sugar with an alcohol.[54] Product alkyl O-glycosides can comprise a mixture of isomers. The α- and β-alkyl O-glycofuranosides tend to be formed kinetically and α- and β-alkyl O-glycopyranosides thermodynamically. Equilibrations regarding ring size and anomeric ratios are difficult to control, so some isomers are difficult to prepare and isolate.
Nuchter et al. studied glycosylation of glucose, mannose, and galactose under microwave heating with alcohols and catalytic amounts of HCl.[55] With glucose and MeOH, methyl O-α-glucopyranoside was formed quantitatively at 140°C after 20 min.
We sought to control the formation of isomers. Acidic mixtures of glucose and MeOH were heated in the MBR at designated temperatures between 70 and 150°C, for 1–15 min, then cooled rapidly and neutralized with NaHCO3. The structures of the starting material and the four products studied are presented in Scheme 3. At 80°C after 2 min (with Amberlyst 15 as catalyst), the methyl β- and α-O-glucofuranosides (formed in 19 and 14% yields respectively) were the only glycosidic products. After 15 min at 70°C, these glucosides comprised 34 and 20% of the product mixture. Lesser amounts of the methyl β- and α-O-glucopyranosides (7 and 5% respectively) had also begun to appear, along with unreacted glucose. At 150°C after 2 min, the thermodynamically most stable products, methyl β- and α-O-glucopyranosides were obtained in 39 and 61% yields, respectively. These results indicated that the rate of conversion of glucose to the methyl O-glucofuranosides was comparable with that for the equilibration of the glucofuranosides to the pyranosides. They also confirmed that although vigorous conditions readily afforded pyranosides as reported, the furanosides could be produced selectively and directly.
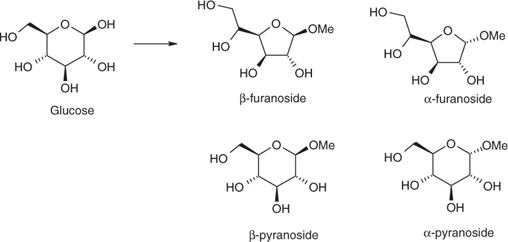
|
With sulfuric acid (at 2.5% w/v with regard to MeOH) as catalyst, at 80°C and with a residence time of 6 min in the microwave zone of the CMR, the methyl β- and α-O-glucofuranosides comprised nearly 80% of the product mixture (52 and 27% respectively). These conditions enabled large-scale preparation of the furanosides. A residence time of 12 min at 80°C reversed the selectivity and methyl β- and α-O-glucopyranosides were obtained in 23 and 54%, respectively.[54]
Solvent-Free Jacobs–Gould Reaction
A key step in the synthesis of quinolone antibacterial agents is formation of a ring possessing an oxo group.[56,57] The vehicle is often a Jacobs–Gould reaction employing elevated temperatures over several hours.[58,59] An environmentally undesirable eutectic mixture of diphenyl ether and biphenyl usually doubles as a diluent and heat transfer medium.
Heat transfer oils for Jacobs–Gould reactions were avoided for the cyclization of enamine 1 to naphthyridone 2, which is a synthetic precursor of nalidixic acid 3 (Scheme 4).[60]
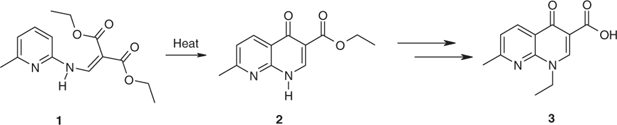
|
Neat methylenemalonate 1 was heated at a range of temperatures between 260 and 300°C. The highest conversion to naphthyridone 2, 44% was obtained at 300°C in 5 min.[60] Extended heating even at lower temperatures resulted in decomposition. Times for conversions up to 40% were plotted against temperature. Extrapolation indicated the shorter periods and higher temperatures required for comparable conversions. These conditions were verified experimentally at temperatures above 300°C. A conversion of 40% or more was only obtained at 290°C and above. Temperatures of at least 370°C and reaction times below 1 min afforded conversions of >80%. When heating was continued for even a few seconds beyond the designated time, however, polymerization occurred. At low heating rates, the unwanted pyrimidinone derivative 4 (Fig. 2) was formed preferentially.
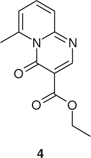
|
In the first example of a solvent-free continuous Jacobs–Gould reaction, flowthrough operation afforded a yield of 86% after 45 s at 385°C, consistent with that for the batch method.[60]
Among other reasons, solvents are used for organic reactions to limit temperature, thermal gradients and adverse effects of oxidation. Conventional teaching has it that high dilution favours intramolecular transformations by retarding competing intermolecular processes. The present work showed that rapid heating to a desired temperature followed by instantaneous quenching afterwards facilitated intramolecular Jacobs–Gould reactions. Without a diluting heat transfer agent, conversion, throughputs, predictability and controllability were high and temperatures were substantially greater than previously employed. The procedure afforded high atom economy,[61,62] was energy efficient, low polluting, offered easy workup and was applied to other quinolones.[60]
Establishing Optimal Conditions
Literature searching for the present report indicated that the term efficient synthesis had rarely been used in journal titles or abstracts before the publication of Silent Spring[3] in 1962. Now, efficient synthesis has appeared more than 9000 times in that context, with ~8000 entries post-dating 1990. Usage of the term appears to parallel the development of green chemistry and to reflect the changing priorities of chemists toward avoiding waste in syntheses.[10] An efficient synthesis would be exemplified by catalysis,[8] a minimum of reactive steps, high atom economy,[61,62] and high yield. An ideal synthesis according to Wender would proceed quantitatively, with 100% atom economy, without waste.[63,64] For the synthesis to be green, the reactants, process and product would need to be hazard-free as well.[10,65]
So far, these concepts have been applied mainly to the assessment of ultimate outcomes. Wastes generated in the course of optimization (e.g. in experiments aiming to increase the yield, decrease the reaction time, or facilitate workup) tend to be discounted as do those associated with the reaction but generated beyond the mixture. Examples include spent heat-transfer oils or coolants, e.g. Me2CO, ice and salt or EtOH/CO2 that are disposed of subsequently, as well as solvents discarded after cleaning of glassware.
As expected with new reactions, in our work, high synthetic efficiencies and yields were seldom obtained the first time. Even with microwave reactors at hand, ~11 experiments usually were required to establish optimal conditions. Although many different optima were possible for any one reaction,[9] optimization involved trial and error and was directed primarily by experience. Even when successful, this process was wasteful of time and resources. Our efforts would not have been unique in that regard.
From another perspective, the rapid uptake of closed vessel reactors (including conventionally heated, pressure-resistant systems) highlights current emphasis on high throughput, a trend that seems more likely to intensify than wane. A potential consequence is that contemporary chemists may become reluctant to repeat traditional, time-consuming processes, even if the yields are respectable. Useful and reliable methods developed over a century and a half could fall into disuse as a result.
Accordingly, we perceived needs for a generic technology that could translate sets of known reaction conditions into alternative, more desirable conditions by guidance rather than through extensive kinetics studies or by trial and error.[41,66] As with time and temperature, yield needed to be treated as a variable reaction parameter instead of as an uncontrollable outcome, a novel concept.
Software for Predicting and Optimizing Yields
Data for ~300 literature reactions and unpublished results from our laboratory were analyzed. Algorithms corresponding to the contours of reaction surfaces were derived and incorporated into an interactive software program that could:
-
Allow conditions for a given reaction to be translated readily into protocols for achieving the same or an improved result in a different time,
-
Be applied to a diverse range of unrelated reactions, and
-
Nominate improved conditions for reactions that proceed poorly or moderately.
As demonstrated by the following worked-through example,[41] the software was iterative:
-
For a thermal reaction, one set of known experimental conditions of temperature, time and either the yield or conversion were required, e.g. after 93 min at 120°C, a conversion of 20% was obtained. Such specific conditions and the corresponding result were termed reference data.
-
In seeking an improved yield or conversion, desired partial conditions were entered into the program, e.g. temperature of 150°C (instead of 120°C as in the reference data) and conversion of 80% (instead of 20%). Reference data from (1) were the basis for calculating the time required. In this example, the time was calculated to be 204 min.
-
Summarizing, the calculation indicated that heating at 150°C after 204 min might afford a conversion of 80%.
-
The subject reaction was carried out at the designated time and temperature. The conversion might have been higher, lower, or in agreement with that calculated.
For data from hundreds of examples analyzed, the correlation coefficient between the expected and the experimental results for the first iteration was ~0.75. Significantly, the data points were relatively evenly distributed about the regression line, supporting the validity of the model.
5. In this example, the conditions in (3) afforded a conversion of 95%, some 15% higher than the conversion sought.
6. The fresh experimental result (i.e. 150°C, 204 min and 95% conversion) constituted new reference data and enabled a second iteration to be performed. The second calculation designated a reaction time of 107 min.
7. The calculated and experimental results were in close agreement.
For the second iteration, the likelihood of a yield or conversion being within ±2% of the calculated outcome was higher than for the first iteration. The correlation coefficient between the expected and experimental results, 0.97, was obtained from more than 300 individual data points.
The above iterative procedure was completed after two calculations and three experiments. Significantly, the ultimate conditions shared no commonality (i.e. different time, temperature, and conversion) and hence no apparent connectivity with those of the reference data. This example was obtained by applying the software to published data for the synthesis of phenyldodecanes by alkylation of benzene over non-zeolitic catalysts, reported by Yadav and Doshi (their fig. 10).[67] The literature reaction was heterogeneous and heated conventionally, so microwaves were not involved.
The software has been applied successfully to diverse, unrelated reactions, saving time, effort, expense, and waste. Optimization typically has been achieved within three experiments. Others now have acknowledged the importance of yield prediction as a valuable enabling tool for planning and selecting synthetic routes.[68,69] Hopefully, future chemists will be able to select a synthetic scheme for a desired product by employing retrosynthetic analysis in combination with predictive calculation of appropriate reaction conditions. An ideal objective would be to settle on the optimal pathway and yield before attempting any practical synthetic work and then to achieve the desired result experimentally, by green chemistry at the first attempt!
High-Temperature Water as a Multi-Tasking Synthetic Medium
In the course of our research, synthetic efficiencies often were sought through consolidation of individual operations into unit steps. Media, reactants, catalysts, and conditions were selected to serve several purposes, sometimes at different stages of a process. Herein, this concept is termed multi-tasking, which is commonly used beyond chemistry and applied to people who perform several different tasks often simultaneously.
With a dielectric constant (ϵ) of 78 at 25°C, water is a poor solvent for most organic compounds at ambient temperature. At 300°C, however, ϵ decreases to 20, a comparable value with that of EtOH and Me2CO at 25°C.[70,71] This enables water to behave as a pseudo-organic solvent at elevated temperature.[9] The ionic product is 1000-fold greater at 240°C than at 25°C, making water a stronger acid and base at higher temperatures. These apparently conflicting properties (decreased polarity yet greater dissociation) confer complex roles that vary with temperature in organic reactions.[52]
When product mixtures in aqueous media are worked up by extraction with organic solvent, each phase becomes saturated with the other. Recycling and disposal become more difficult as a result. Environmental benefits gained through using water as the reaction medium in the first place are lost in the workup. This aspect tends to be overlooked by advocates of water as a green solvent, a sound reason for considering the synthetic cycle as a whole.
The problem was alleviated by employing hydrophobic resins with high surface areas (~800 m2 g–1) for concentration and isolation of the products from aqueous media.[53] Organics, typically ~40 mg per g, were retained on the resin and subsequently could be desorbed. EtOH was the preferred solvent in that regard. It has appropriate solvent properties, low toxicity, is readily recyclable by distillation and is both a renewable and a biodegradable resource. Also, it forms an azeotrope with water, avoiding needs for inorganic drying agents that would add to waste. Other advantages of non-extractive processes include convenience, high throughput, and decreased effluent owing to ready disposal of the spent water. The resin and the solvent used for desorption could be recycled.
An example of the combined use of microwave heating, water as a multi-tasking solvent or medium, followed by non-extractive workup was the preparation of 2,3-dimethylindole from PhNHNH2 and MeCOEt (Scheme 5). This was the first example of water as a medium for Fischer indole synthesis.[9] The process did not require a preformed hydrazone or added acid. Only one reactive step was involved and the resin technique (unpublished for this example) could be employed to isolate the product. Multi-tasking roles of water varied according to the temperature and included its serving as a solvent, medium, and acidulant in the synthetic step (at high temperature) and as a polar medium that facilitated isolation of the product onto a hydrophobic resin (at ambient).
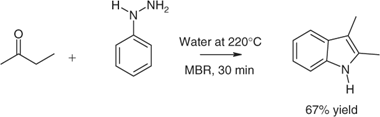
|
Indole cannot be obtained easily from PhNHNH2 and MeCHO by Fischer synthesis. Traditionally, indole-2-carboxylic acid has been prepared from phenylhydrazine and a pyruvate ester, followed by hydrolysis. Methods for decarboxylation of indole-2-carboxylic acid to indole included heating with copper salts, in heat-transfer oils, in glycerol, quinoline, or in 2-benzylpyridine. In a green microwave-assisted reaction, however, decarboxylation of indole-2-carboxylic acid occurred quantitatively in water at 255°C within 20 min (Scheme 6).[9]
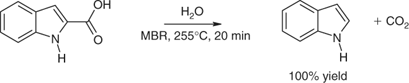
|
It did not proceed if the indole-2-carboxylic acid was deprotonated.[72] The presence of base inhibited the decarboxylation but facilitated hydrolysis of ethyl indole-2-carboxylate. Thus, the conditions could be controlled to produce either the acid or indole in high yield and selectivity from the ester, by carefully manipulating the equivalents of base present.[72]
High-temperature water was effective for other reactions including hydration of olefins, elimination of alcohols, isomerization, nitrile hydrolysis, as well as ortho-Claisen, Rupe and Meyer–Schuster rearrangements.[52] Literature methods for corresponding reactions with alternative media tended to require orders of magnitude higher levels of acid or base and sometimes high-boiling organic solvents.
Inorganic salts form the bulk of waste from conventional industrial chemical reactions.[7] Often produced by neutralization of acidic or basic solutions, they enter soil and ground-water, rendering them unusable for farming or drinking. By lowering the pH of atmospheric moisture, they contribute to acid-dew and acid-rain, which degrades vegetation as well as engineered structures. As originally pointed out by Sheldon,[7] minimization of salt formation is essential in working up reactions. Because reactions in high-temperature water only required low concentrations of mineral acid or base to proceed satisfactorily, commensurately low concentrations of salt were generated in subsequent neutralization steps. This represented a significant technical and conceptual advance toward green protocols.
In summary, conditions for several established reactions have been improved by multi-tasking roles of water. Improvements included reducing the number of steps through tandem reactions, minimizing salt formation, lowering reaction time, decreasing the amount of catalyst added (in some cases to zero) and avoiding solvent extraction during workup.
Although high-temperature water was widely considered impractical as a solvent or medium for organic synthesis when our studies began, there is now great interest in its applications,[73,74] including under microwave conditions.[75] Obvious advantages include low cost, negligible toxicity and safe handling and disposal. Recently, a commercial microwave batch system was used to perform aqueous reactions safely at 300°C and 8 MPa. At higher temperature, Kappe et al.[76] found improved conditions for some of our published reactions, including that for 2,3-dimethylindole, where we had used 220°C (Scheme 5). Somewhat surprisingly, the authors introduced the term ‘near-critical’ instead of using ‘high-temperature’, as we had many years earlier, to describe the condition of water above its atmospheric boiling point, but well below its critical temperature (Tc). As 300°C is more than 70°C below, i.e. well below the Tc of water, the use of ‘near-critical’ appears to be somewhat akin, in relative terms, to referring to water at 80°C as ‘near-boiling’ when it would be normally described as ‘hot’. Given the changes of state associated with the boiling, critical and freezing points, terminology such as ‘near-critical’ appears to be as inappropriate as referring to water at ambient as ‘near-freezing’.
A ‘Distillable’ Protic Ionic Liquid
Ionic liquids commonly are defined as salts with melting points below 100°C and composed totally of ions. Those that are liquid under ambient conditions are sometimes referred to as room-temperature ionic liquids. They usually have no detectable vapour pressure at atmospheric pressure and can dissolve a wide range of inorganics and organics.[77] These properties have afforded a range of applications, including as replacements for volatile organic solvents.[78–80]
Varma and Namboodiri were first to demonstrate benefits of microwave heating for the preparation of ionic liquids.[81] Soon afterwards, Deetlefs and Seddon prepared 1-alkylpyridinium, 1-alkyl-3-methylimidazolium, 1-alkyl-2-methylpyrazolium, and 3-alkyl-4-methylthiazolium ionic liquids that way.[82] Reaction times were dramatically reduced compared with conventional heating. Syntheses were performed in sealed or open vessels on scales ranging from 50 mmol to 2 mol. The molar excess of haloalkane previously required for conventional thermal ionic liquid syntheses was significantly reduced.
Several reports of microwave-assisted synthesis of ionic liquids have since appeared, including a comparison of the efficacy of various commercial reactors for scaling-up.[83] Opportunities for new applications based on synergies between microwave technology and ionic liquids were reviewed recently by Leadbeater and Torenius.[84] They arise mainly from the following:
-
Ionic liquids are very effective microwave absorbers, so heating can be rapid.
-
Ionic liquids usually have no effective vapour pressure and can be used at moderately high temperature and at atmospheric pressure.
These aspects are under investigation in several laboratories.[84] On some occasions though, the negligible volatility of ionic liquids can complicate purification and isolation processes. Removal of the ionic liquid from the product can be difficult if, as is often the case, both are involatile and mutually soluble.
To access advantages of ionic liquids, yet facilitate workup, the use of N,N-dimethylammonium N,N-dimethylcarbamate (dimcarb)[85,86] was investigated. The pure carbamate is a 1:2 crystalline adduct of CO2 and Me2NH respectively, both of which are gases at room temperature. Typically, a mixture of species is formed from the gases in a spontaneous and exothermic reaction (Scheme 7).
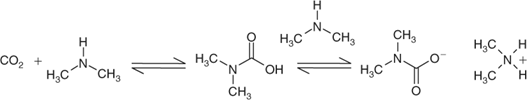
|
The molar ratio usually is nearer 1:1.8 and under those circumstances dimcarb exists as a relatively stable liquid up to 50°C. It can be produced in bulk, readily and inexpensively, and has substantial ionic character.[87] Some salts, e.g. LiCl, NaCl, NaBr, KCl, and KI, dissolve in it at levels between 2 and 5% w/v.[85,86] Although lower than that of water and higher than that of hydrophobic organic solvents, such polarity is comparable with that of ionic liquids.[88] At 60°C, dimcarb undergoes reversion, facilitating isolation of the product. The CO2 and Me2NH can be condensed and re-associated and dimcarb recovered for re-use. Hence, in our work dimcarb was recognized as a self-associated, ‘distillable’ protic ionic liquid.[89]
A New Platform for Phenol-Formaldehyde-Based Chemicals and Materials
Phenolic derivatives and formaldehyde undergo coupling reactions to produce resins that have been used widely in the cookware, plywood, particleboard, abrasives, and adhesives industries for up to a century.[90] Calixarenes, cyclic oligomers produced as by-products in phenol/formaldehyde reactions, were recognized ~60 years ago.[91,92] They constitute a field on their own, with applications including ion scavenging, chromatography, controlled release, and enzyme mimicry.
Vapours of phenol can cause respiratory irritation and headaches and skin exposure can cause burns, liver damage, and an irregular heart beat.[93] Formaldehyde is a suspected carcinogen.[94] Thus, although the products of phenol-formaldehyde chemistry have high demand, the chemistry toward them employs environmentally undesirable starting materials and empirical methods that hamper the introduction of structural diversity. Consequently, a new, green, technology platform was conceived. It was based on the use of cyclohexanone and benzaldehyde derivatives as building blocks instead of phenol and formaldehyde.[95] In situ formation of dienone species was the key process for either chain extension or macrocyclic ring closure. The cyclohexa-2,6-dienones so formed were set up for isoaromatization to unmask phenolic systems.[96] With increased conformational flexibility and bearing structural elements of calixarenes and crown ethers, the macrocyclic products were named Horning-crowns (see example in Scheme 8). Some examples exhibited solvent-dependent and switchable conformations that could be incorporated into the molecules by design.[97,98] Linear oligomers were also produced.[99,100]
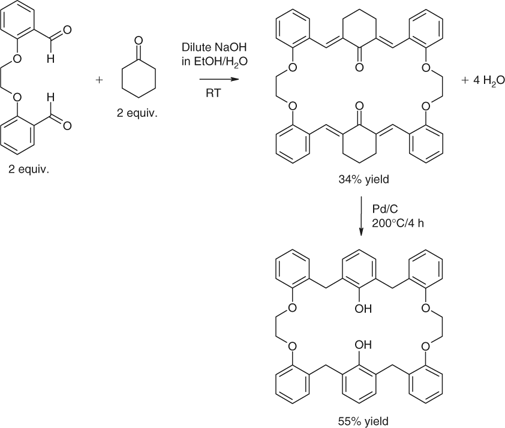
|
Direct Preparation of 2-Benzylidenecycloalkanone Derivatives in Dimcarb
Mono-2-arylylidene derivatives of cyclic ketones, particularly cyclohexanone, were required as building blocks for the dienone-phenol platform outlined above. Direct preparation from the parent ketones and non-enolizable aryl aldehydes by Claisen–Schmidt condensations was not straightforward, however.[95] Literature reports indicated that typically, diadducts such as 2,6-dibenzylidenecyclohexanone or 2,5-dibenzylidenecyclopentanone are formed exclusively, even when the molar ratio of starting aldehyde to ketone is substantially below 1:1 as shown in Scheme 9.
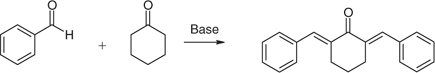
|
Traditionally, mono-2-arylylidene cycloalkanones have been formed in two steps: aldol addition, which is readily reversible, followed by a separate elimination. In dimcarb, the desired products were obtained from the corresponding aldehyde and ketone in one step without resort to protecting groups (Scheme 10). Selectivity and yields were usually high. The pathway involved a Mannich reaction with release of water followed by elimination of dimethylamine.[95]
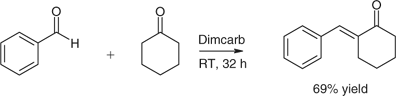
|
Cascade Reaction for Aniline Derivatives
The reaction of benzaldehyde with cyclohexenone (instead of a cycloalkanone) in dimcarb afforded 2-benzyl-N,N-dimethylaniline as a significant by-product. Optimization afforded a general one-step multi-component preparation of N-monosubstituted or N,N-disubstituted aniline derivatives.[101] The reactants comprise an aldehyde without an enolizable carbonyl function, cyclohex-2-enone or a derivative thereof and an amine, which may be primary or secondary. The simplest cases involve monoaldehydes and monoamines (see Scheme 11 for examples featuring benzaldehyde with primary or secondary amines). With a dialdehyde or diamine, the process can occur twice, so great complexity can be introduced in one step.
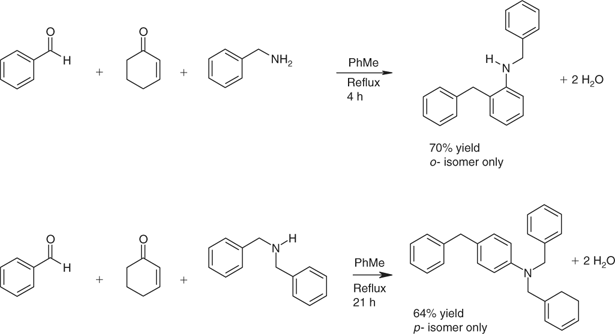
|
The cyclohexenone molecule undergoes extensive transformation, triggered by the formation of an enimine. Water (2 equiv.) is the major by-product and atom economies are typically ~90%. This cascade offered opportunities for the introduction of combinatorial diversity into the new secondary and tertiary anilines, while satisfying many of the principles and objectives of green chemistry.
Regioselectivity usually was high when careful attention was paid to the conditions. Catalysts and rates of addition influenced the outcomes. Primary amines mainly afforded 2-substituted N-substituted anilines and secondary amines tended to favour formation of 4-substituted N,N-disubstituted anilines. A diverse range of aldehydes was employed, extending to heterocyclic compounds such as furfural and aryl dialdehydes. Benzene-1,3-dicarboxaldehyde with cyclohexenone and benzylamine gave 2,2′-[1,3-phenylenebis(methylene)bis(N-benzylaniline)]. The yield (65%) was respectable in light of the potential for competing processes and the number of transformations involved in the assembly of a somewhat complex molecule in one step (Scheme 12).

|
The new reaction was convergent and predictable, regardless of the diverse range of starting materials employed. It was employed combinatorially to establish libraries for active compound discovery and for ligands. Participating amines included primary examples, e.g. benzylamine, aniline, 2-aminomethylpyridine, methyl aminoacetate, tryptamine, or secondary, e.g. di-(2-methoxyethyl)amine, morpholine, amphetamine, and di-n-butylamine.[101]
Potential Impacts of Deep-Sea Disposal of CO2
Not all environmental implications of chemicals result from chemical laboratories or chemical manufacturing. Perhaps the most topical example concerns the effects of increasing atmospheric concentrations of greenhouse gases such as CO2 and CH4 on climate. Burning of fossil fuels is the predominant means of power generation globally. A concentrated form of carbon (coal or natural gas) is diluted by two to three orders of magnitude during combustion (formation of CO2 in air) and by a further three to four orders of magnitude on atmospheric release.[102] Increasing energy usage has enhanced the atmospheric concentration of CO2 from 280 ppm at the beginning of the 19th century to ~370 ppm presently. The ecosystem is unable to process the anthropogenic gas quickly enough for a steady state to be attained,[103] thereby creating conditions for global warming.[104]
Apart from photosynthesis, where green plants utilize the gas as a starting material, CO2 is sequestered naturally by slow diffusion into seawater followed by formation of stable carbonate salts that fall to the ocean floor.[102] Over 30 years ago, Marchetti[102] and Nordhaus[105] suggested that atmospheric CO2 levels could be stabilized and carbon sequestration could be short-circuited by collecting the gas at its source (power stations), compressing it and pumping it onto the seabed. The proposal became the subject of several studies,[106,107] including in collaborations at international government to government level. It gained considerable support and implementation appeared to be imminent ~5 years ago.
In contrast with the natural process outlined above for oceanic sequestration of CO2, deep-sea disposal of the compressed gas would produce pockets of liquid CO2 or clathrates. It had the potential to afford biphasic systems with properties that rarely occur naturally.
Supercritical CO2 has been widely explored as a green solvent and has found laboratory uses and industrial applications in decaffeination of coffee, dry cleaning, and for extraction of dyestuffs.[108] Such aspects did not appear to have been taken into account by the proponents of deep-sea disposal of CO2. We considered that such disposal of CO2 could affect the nature and distribution of organics in seawater by mechanisms including sequestration, reactions involving CO2 and by localized increased acidity of seawater.[109] Extraction and concentration of natural products from seawater into the organic phase could promote processes that would not normally occur, e.g. possible reaction of CO2 with amines to produce carbamates. Among other alternatives, potentially harmful organic compounds that otherwise would be hydrolyzed in seawater could be sequestered and stabilized in the CO2 phase instead of being degraded.
Our preliminary study employed 1,2-epoxides with CO2/saltwater mixtures.[109] Epoxides have biological roles and undergo ring-opening by substitution, addition, rearrangement, and insertion, including by CO2: they react under a range of conditions and largely by established mechanisms along competing pathways.[110] Although the epoxides tended to be relatively stable in salt water, they degraded rapidly in CO2-saturated saltwater and in biphasic CO2-saturated saltwater/liquid CO2 systems. Chlorohydrins, potential alkylating agents, were significant products. Further experiments indicated that increased acidity of the saltwater through the introduction of CO2 had a role in the enhanced reactivity of the epoxides and the products formed (Scheme 13).
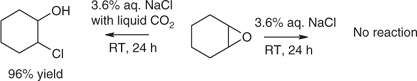
|
Before our work, the origins of β-halohydrins from terrestrial and aquatic sources had been ascribed to either combustion of organics in the presence of halides or halogens or to biosynthesis involving haloperoxidases.[111,112] Addition of the elements of HBr, HCl, and HI (derived from the corresponding sodium salts in water) to epoxides in the presence of CO2 now could be considered as well, particularly for metabolites of corals and coralline microalgae[113] that inhabit seawaters containing high levels of CO2. Hence, our results were not only cautionary with regard to possible environmental implications of CO2 disposal in seawater, a proposal that now seems to have been abandoned, they also indicated that saltwater/liquid CO2 systems could have useful synthetic applications at least with regard to opening of epoxides.
Concluding Remarks
From tentative beginnings in the late 1980s, microwave chemistry has developed into a significant field. More than 3000 research papers have appeared, as have numerous reviews and, since 2002, several monographs.[44,114–116] Commercially available closed-vessel reactors based on our designs and concepts have broadened the range of accessible temperatures and have helped to speed up and automate synthetic chemistry globally. They are employed extensively and routinely in chemical discovery, organic synthesis, and medicinal chemistry and have found niche applications in the production of intermediates, flavours and fragrances, specialty chemicals and pilot-scale manufacture.
Most peer-reviewed chemical journals no longer accept reports on microwave reactions unless sufficient experimental detail is provided to enable others to replicate the conditions. Conferences devoted to the field feature plenary and keynote lectures by a new wave of microwave synthetic chemists and process specialists, who were either not engaged in the field a decade ago or perhaps were just starting.[44,114] Through substantial and extensive contributions that lie beyond the scope of the present personal account, such researchers have taken the field into new and exciting directions.
Enabling tools and methods were developed for activities now encompassed by green chemistry. The approach was demonstrated through the development of a technology platform consisting of structurally diverse oligomers, macrocycles, and rod-like molecules that could supplement, and in some cases, may replace some of those available through phenol-formaldehyde chemistry. Products were synthesized with ‘male’ and ‘female’ building blocks (analogous to plumbing fittings) connectable by Claisen–Schmidt or Mannich type reactions. In addition, principles of green chemistry were invoked to provide experimental results to counter a proposal for accelerating sequestration of atmospheric CO2 by deep-sea disposal as liquid.
Although green chemistry is accepted much more widely now than in 1988, somewhat surprisingly it still appears to be regarded by some chemists as a ‘soft’ science. Principles of green chemistry, however, rule out the use of toxic or hazardous materials, reagents and solvents, which traditionally constituted a large proportion of the synthetic chemist’s arsenal.[10,65] The comparatively limited range of such materials and methods that are hazard-free and waste-free makes it considerably more challenging to perform synthetic chemistry in green ways than by conventional means. This indicates that green chemistry is not a ‘soft’ science and that conversely, for many it may still be too hard! Hopefully, an ever-improving array of new, convenient clean technologies and methods will encourage increasing numbers of organic chemists to join this endeavour and to contribute toward its success.
Acknowledgements
The author wishes to thank his coauthors in the articles cited for their dedication and perseverance in bringing much of the research to fruition. Professor D. W. Cameron, Dr P. J. Williams and Professor K. R. Seddon are thanked for support and encouragement far exceeding any call of duty. Milestone (Italy) is thanked for helping to transform microwave dreams into commercial reality. Gabriela Adamova is thanked for producing the figures for the present work.
[1]
[2]
A. A. Blumberg,
J. Chem. Educ. 1994, 71, 912.
|
CAS |
|
CAS |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
|
CAS |
|
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
|
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
|
CAS |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
|
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
|
CAS |
|
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
|
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
|
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
|
CAS |
|
CAS |
|
CAS |
|
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
(verified 14 August 2008).
[94]
[95]
L. T. Higham,
U. P. Kreher,
C. L. Raston,
J. L. Scott,
C. R. Strauss,
Org. Lett. 2004, 6, 3257.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
|
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |

[114]
[115]
[116]