

Mechanistic considerations for transmetalation at nickel(II) and palladium(II) complexes: towards improved catalysis
Nicholas S. D. Solomon A * and Sinead T. Keaveney B *
A * and Sinead T. Keaveney B *
A
B

Dr Nicholas Solomon received his PhD in 2025 from Macquarie University in Sydney, Australia, having studied organometallic reaction mechanisms under the supervision of Dr Sinead Keaveney. Following this, he joined the Dementia Research Centre at Macquarie University as a post-doctoral researcher in the group of Ole Tietz. His work now focuses on developing small molecule and peptide-based therapeutics, as well as synthetic strategies towards these applications. |

Dr Sinead Keaveney received her PhD in 2016 from UNSW Australia, in the field of physical organic chemistry under the supervision of Assoc. Prof. Jason Harper. Following a post-doctoral position at RWTH Aachen University, Germany, with Prof. Franziska Schoenebeck, and a research fellowship at Macquarie University, Australia, she joined the University of Wollongong in 2021 as a lecturer. Her research program is focused on developing new synthetic methodology through combined experimental, computational and mechanistic studies. |
Abstract
Palladium and nickel catalysed cross-coupling reactions are used extensively in organic synthesis, with state-of-the-art cross-coupling methodology enabling access to diverse building blocks in an efficient and selective manner. In recent years, there has been growing interest in using nickel-based catalysts for cross-coupling reactions, in place of the more commonly used palladium catalysts. This is motivated by both the unique reactivity that can be accessed when using nickel catalysts, and due to nickel being cheaper and more earth abundant than palladium. Despite these advantages, larger catalyst quantities and higher reaction temperatures are often required when using nickel, increasing cost and environmental impact. As such, in order to design more efficient nickel-catalysed cross-coupling reactions, a greater understanding of how these catalysts operate is required. In this review, mechanistic studies on the key transmetalation step of typical palladium and nickel-catalysed cross-couplings are discussed, with a focus on how catalyst structure affects the efficiency of transmetalation at palladium(II) and nickel(II).
Keywords: cross coupling, density functional theory, DFT, kinetics, mechanism, nickel catalysis, palladium catalysis, transition state, transmetalation.
Introduction
Cross-coupling reactions are one of the most widely used approaches to constructing C–C and C–heteroatom bonds, due to the efficiency, functional group tolerance and method mildness that this synthetic strategy offers. For the past 50 years or so, palladium catalysts have commonly been used to achieve efficient cross-coupling (high product yield when using low catalyst loadings), though in recent years there has been increased interest in developing nickel-catalysed cross-coupling strategies.
This has been motivated in part by cost, with palladium being substantially more expensive than nickel owing to its very low natural abundance in the Earth’s crust (15 µg kg–1 compared to 84 mg kg–1 for nickel).1 This difference in abundance is routinely used to promote nickel catalysts as more sustainable than their palladium counterparts, though in practice, this must be weighed against the larger catalyst quantities and higher reaction temperatures often required by nickel catalysts.2 At present, nickel catalysts have not yet reached their full potential, with a major challenge being the difficulty in predicting reactivity.
For example, unlike palladium, nickel can readily access +1 and +3 oxidation states – this can hamper the prototypical 0/+2 cross-coupling cycle, cause catalyst deactivation and frustrate efforts to optimise reactions with the aid of NMR spectroscopy, as these species are paramagnetic.3 However, this departure from well established reactivity provides opportunities to develop new reactions – a major advantage to nickel catalysts and current point of interest to many researchers. A similar duality extends to the propensity for nickel catalysts to undergo radical processes, their affinity for unsaturated bonds and their reactivity towards less reactive C–Cl, C–F and C–O bonds. These features can lead to poor selectivity, poor functional group tolerance and ultimately poor yield if traditional cross-coupling transformations are desired, though they can all be of great utility in developing new reactions.3
With this considered, the key to unlocking the true potential of nickel catalysts, both in terms of traditional cross-coupling and novel reactivity, is a greater understanding of how they operate mechanistically. Improved selectivity, higher yields, milder reaction conditions and lower catalyst loading requirements can be achieved through mechanistically informed reaction design.
To this end, when considering typical cross-coupling catalysis (through Pd0/II or Ni0/II), three mechanistic steps must be examined: oxidative addition, transmetalation and reductive elimination. Notably, the reductive elimination step is typically rapid,4 especially for nickel catalysts,5,6 and as such, when designing nickel-catalysed cross-coupling reactions, tuning the catalyst to promote oxidative addition and transmetalation generally becomes more important. Of these steps, oxidative addition has been extensively studied,7 but significantly less rate and mechanistic data are available regarding transmetalation, especially at nickel.
Studies on transmetalation at palladium and nickel
The comparatively sparse data available concerning transmetalation at PdII or NiII complexes is the result of several factors: firstly, when studying transmetalation, it is generally true that pre-prepared complexes of the form [(Ln)M(R)(X)] must be synthesised, purified and characterised before kinetic analysis can commence. By contrast, for studies on oxidative addition, commercially available species such as Pd(PPh3)4 and Ni(PPh3)4 can be used to gain a great deal of insight.8–12 Compounding this difficulty, catalytically relevant complexes of the form [(Ln)M(R)(X)] are often intrinsically unstable; for a catalyst to be efficient, it must be sufficiently unstable to avoid it from acting as a thermodynamic sink in the catalytic cycle.
Further complicating matters, although the prototypical cross-coupling reaction (oxidative addition, transmetalation, reductive elimination) is presented as a neat cycle, in practice, numerous catalyst decomposition pathways may also be present. For nickel, this often involves the generation of paramagnetic NiI species, making kinetic analysis by NMR spectroscopy difficult. As a result, most of the kinetic and mechanistic data pertaining to transmetalation in cross-coupling reactions have focused on palladium. In this review, a summary of experimental studies related to transmetalation at palladium(II) and nickel(II) complexes is presented, with a particular focus on the mechanism and reactivity trends determined for transmetalation at nickel complexes.
Transmetalation with organo-tin reagents – the Stille coupling
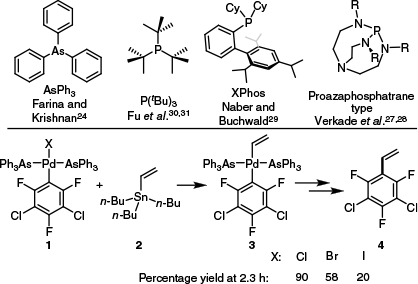
Regarding transmetalation in Pd-catalysed Stille coupling reactions, extensive studies have been reported by Espinet,13–19 Stille20–23 and Farina.24–26 It has been determined that transmetalation is more rapid when weakly coordinating or bulkier monodentate ligands are used (Scheme 1, top),24,27–31 and transmetalation follows the reactivity trend I > Br > Cl (Scheme 1, bottom).13 This experimentally derived trend was further substantiated by a computational study examining transmetalation at Pd complexes bearing PH3 ligands,32 with transmetalation at [(PH3)Pd(Ph)X] by trimethyl(vinyl)tin calculated to occur with activation energies of 57, 75 and 93 kJ mol–1 for X = Cl, Br and I respectively. Beyond the typical halogens, triflate complexes of the form [(L)2Pd(Ar)X] (L = PPh3 or AsPh3; Ar = 2,4,6-trifluoro-3,5-dichlorophenyl) undergo transmetalation more quickly than their chloro counterparts.14
Ligands that are particularly competent in the Stille coupling (top) and data obtained by Casado and Espinet showing the rate of transmetalation at [(LL)Pd(Ar)X] complexes 1 that differed in X (Cl, Br or I).13,24,27–31
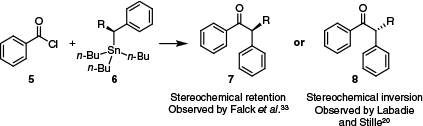
Importantly, until the early 2000s, considerable ambiguity surrounded the nature of the transition state for this transmetalation reaction – a consequence of conflicting reports from the groups of Labadie and Stille20 and Falck et al.33 when examining the reactivity of chiral benzyl stannanes 6 (Scheme 2). Falck et al. observed retention of configuration to produce compound 7,33 whereas Labadie and Stille reported inversion of configuration to compound 8.20
The inversion or retention of stereochemistry that may be observed for the Stille coupling, depending on the conditions used.20,33
These anomalous results were later reconciled by Casado and Espinet, who proposed two competing mechanisms through which transmetalation could occur: a cyclic mechanism or an open mechanism.13,14 The derivation of the cyclic mechanism came from kinetic analysis, which found transmetalation to be 1st order with respect to stannane and –1st order with respect to the ligand. Although the latter observation, coupled with the fact that transmetalation occurred more rapidly for weakly coordinating ligands (such as arsines),24,32 had previously been attributed to the need for formal ligand dissociation ([(L)2PdRX] forming [LPdRX]), Casado and Espinet proposed an associative mechanism in which transmetalation and ligand dissociation occurred concurrently. Importantly, this was consistent with the reaction being 1st order in stannane, whereas a mechanism involving an initial rate-limiting ligand dissociation step would necessarily preclude a non-zero reaction order in stannane. Overall, a mechanism involving a cyclic intermediate was proposed (Scheme 3), which would result in retention of stereochemistry. Experimental validation of the cyclic pathway was later provided by Santos and co-workers, who identified the cyclic intermediate through mass spectrometry.34
The cyclic SE2 pathway proposed for transmetalation during certain Stille coupling reactions at PdII.
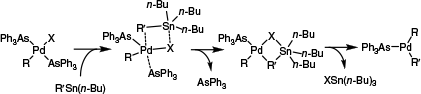
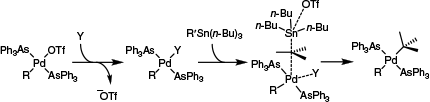
By contrast, while later examining Stille cross-coupling reactions involving organo-triflates, Casado and Espinet recognised the need for a second mechanism to explain reactivity in the absence of a competent bridging ligand (such as halogens). In particular, it was recognised that triflate dissociation from [(AsPh3)2Pd(Ar)OTf] followed by additional ligand coordination to generate the cationic complex, [(AsPh3)3Pd(Ar)]+, was favourable at elevated temperatures in THF (K = 2.3 at 50°C; corresponding to ΔG = –2 kJ mol–1).14 This equilibrium approximately corresponded to computational data for triflate dissociation from the analogous complex, [(PH3)2Pd(Ph)OTf], in THF (ΔE = +2.9 kJ mol–1; assuming ΔS for ligand exchange is small, ΔG ≈ ΔE).15 Similarly, in the presence of a strongly coordinating and bridging halide (such as Cl), use of highly coordinating solvents (such as HMPA or NMP), or an excess of ligand allowed halide displacement to occur.14,16 As such, under certain conditions, cationic complexes are relevant catalytically. Additionally, it should be noted that these species are highly electrophilic, especially in the presence of weakly donating ligands like arsines. Combined, these effects enable transmetalation to occur through an SE2 electrophilic cleavage mechanism with inversion of stereochemistry (Scheme 4).
The open SE2 pathway proposed for transmetalation during certain Stille coupling reactions at PdII. Y, solvent or additional ligand; R′, tBu for illustrative purposes.
Beyond the nature of the catalyst, it is also important to note that qualitatively similar transmetalating agents (e.g. vinyl- v. ethynyl-stannanes) can differ substantially in their reactivity. One of the first studies illustrating this effect came from Labadie and Stille, who reported the order of reactivity to be Ph–C≡C > alkyl-C≡C > Ph–C═C > vinyl > phenyl > benzyl > CH2OCH3 > CH3 > butyl, for a model cross-coupling reaction between benzoyl chloride 5 and substituted stannanes 9 (Scheme 5).20 Additionally, when a range of substituted benzyl groups was studied, it was determined that electron-withdrawing groups accelerated transmetalation (P = 1.2).20 This was in accordance with the rate of acid hydrolysis of phenyl tin reagents,35 which progresses through an SE2 type mechanism.36
A summary of the rates at which different tributyl(organo)tin reagents underwent transmetalation in a model Stille reaction.
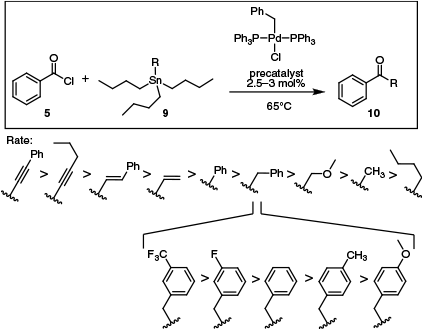
Taken together, these data suggest that the coordinating ability of the organic group is important, with better η2-ligands more readily able to coordinate to the metal centre, as is required prior to transmetalation in the strict sense (Sn–C bond breakage and Pd–C bond formation). In addition, electron-deficient R groups polarise the C–Sn bond, making cleavage easier and accelerating transmetalation.
Although the above information describes palladium-catalysed Stille couplings, a very limited amount of data are available for the analogous nickel-catalysed reaction. In the 1990s, Hill’s group observed that [(PPh3)2NiCl2] and [(dppf)NiCl2] both performed poorly in the coupling of aryl-mesylates with PhSn(n-Bu)3, achieving fewer than three turnovers.37 They attributed the low activity to slow transmetalation, although under similar conditions, palladium catalysts also performed poorly.
A few years later, some success was attained by Shirikawa and co-workers.38 They found Ni(PPh3)n to be a suitable catalyst for Stille couplings in which chloro- and bromoarenes were coupled with active organostannanes.38 This study was supplemented with another publication in which aryl chlorides, bromides, iodides and triflates were found to be suitable substrates.39 Notably, some insight into the order of this nickel-catalysed reaction could be gained, with 4-(trifluoromethyl)iodobenzene 11 (for which oxidative addition would be very rapid and almost certainly not rate determining) and stannane 2 coupling at a rate approximately inversely proportional to ligand loading (product 12 yield after 3 h).39 Analogous to palladium catalysts, excess ligand inhibited the reaction (Scheme 6).
The inhibitory effect of added ligand on product yield. This was consistent with the trend observed for the same reaction conducted using an analogous Pd catalyst.
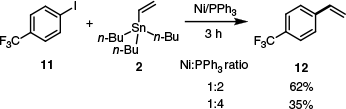
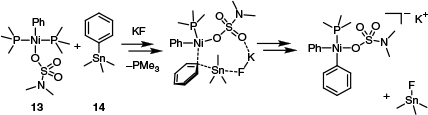
Recently, Neufeldt’s group successfully demonstrated a Ni-catalysed Stille coupling using moderately bulky, monodentate phosphines that featured a combination of alkyl and aryl groups (PPhEt2, PPhMe2, PPh2Me).40 Illustrating the capacity for nickel to afford new reactivity, aryl dimethylsulfamates could be coupled with a range of organostannanes in the presence of potassium fluoride.40 Based on density functional theory (DFT) data, Neufeldt’s group suggested the role of KF was to aid transmetalation by forming an extended cyclic structure in which fluoride activated the stannane 14 by an outer-sphere mechanism (Scheme 7).
Transmetalation with organo-silicon reagents – the Hiyama coupling
The majority of reported mechanistic studies for transmetalation with silanes have involved palladium, with limited data available for transmetalation between organosilanes and nickel. In general, the factors that influence the rate of transmetalation with silanes are comparable to those observed with stannanes. Notably, however, organosilanes are much less reactive than their stannane counterparts, with a reactivity series: Sn > Ge > Si.41 For practical purposes, this means it is necessary to use highly activated organo-silicon species, or to use activating additives to promote transmetalation.42–45 In recent years, additive-free Hiyama-type cross-coupling reactions have been reported, which have relied on the generation of Pd–F or Ni–F intermediates, which can react directly with the organosilane.46–49
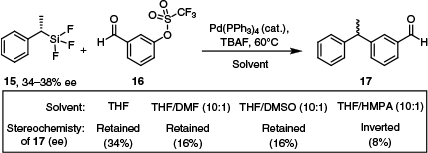
In terms of similarities between silanes and stannanes in cross-coupling reactions, the same cyclic and open SE2 transition states have been invoked for both reagent types based on similar stereochemical observations (Scheme 8).20,33,50 Comparable to observations regarding the Stille coupling, with Hiyama couplings, coordinating solvents favour halide displacement, the open mechanism and stereochemical inversion. The opposite is true in less coordinating solvents, which favour a cyclic SE2 mechanism and retention of stereochemistry. This was elegantly demonstrated in an examination of the stereochemical scrambling of the coupling product 17.50
Stereochemical retention, partial retention and inversion observed for a model cross-coupling reaction between 15 and 16 in different solvents.
Similarities between silanes and stannanes in transmetalation reactions have also been observed computationally. Some 30 years after the initial publication of Hiyama et al. regarding the addition of fluoride to silanes in the context of cross-coupling reactions, the same author investigated a prototypical silane transmetalation by DFT to elucidate the role of the fluoride additive.51 It was found that pre-activation of the silane to generate a hypervalent, anionic adduct was not kinetically possible (Fig. 1, left). By contrast, halide exchange (iodide to fluoride) followed by a cyclic transition state, as well as an open SE2 type mechanism, were both kinetically viable (ΔE‡ = 105 and 69 kJ mol–1 respectively) (Fig. 1, right). The same was true when hydroxide was modelled in place of fluoride. This study suggests that, despite the need for an additive, transmetalation with organosilanes progresses by the same mechanisms (cyclic and open SE2) as organostannanes.
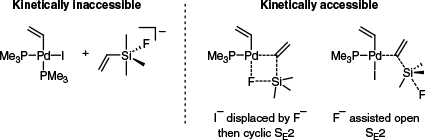
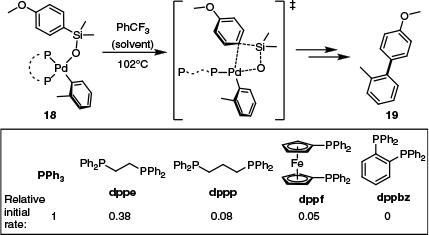
The finding regarding the role of hydroxide was consistent with the work of Denmark’s group, who reported that silanols and related species can undergo transmetalation by an SE2-type transition state. They also found that ligand selection had a pronounced impact on the rate of transmetalation with silicon-based reagents.52 Among the bidentate ligands examined, the trend of reactivity for a model transmetalation reaction was found to be dppe > dppp > dppf ≫ dppbz (Scheme 9). This was generally consistent with the structural rigidity of each ligand, as well as their propensity for partial dissociation. Furthermore, a complex bearing two mono-dentate ligands, PPh3, was the most reactive of the series (Scheme 9), again mirroring trends reported with stannanes.
SE2-type transmetalation with silanolates investigated by Denmark’s group,52 and the trend in reactivity associated with ligand choice that was observed.
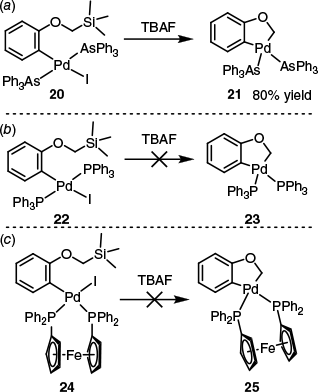
A similar finding came from Echavarren’s group,53 who examined transmetalation using an intramolecular system. Mirroring the Stille coupling and the work of Farina and Krishnan,24 they found that complex 20, bearing AsPh3 ligands, was particularly reactive towards transmetalation (Scheme 10a). By contrast, the analogous PPh3 22 and dppf 24 ligated complexes were unreactive (Scheme 10b, c).53 Again, highlighting the rate enhancements observed with monodentate ligands, Ju and co-workers found PPh3 to be more effective than bidentate dppf or dppe in the palladium-catalysed Hiyama coupling of arylbromides with trimethoxy(phenyl)silane,54 whereas comparable results were also obtained by Denmark and Wu, who found PCy3 and AsPh3 to be particularly effective in the palladium-catalysed coupling of aryl(halo)silacyclobutanes with aryl iodides.55
Intramolecular transmetalation occurred in the presence of (a) AsPh3 (complex 20), but not (b) PPh3 or (c) dppf (complexes 22 and 24 respectively).
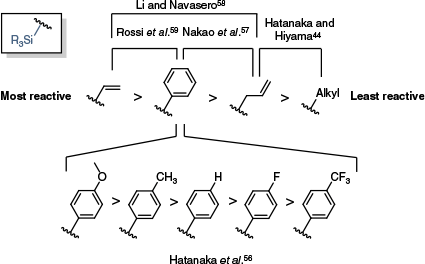
Although little data exist in the way of direct comparisons or a reactivity series for differently substituted silanes, it is possible to gain some qualitative insight into reactivity trends based on a collection of reported studies, which have been summarised in Fig. 2.44,56–59 Of note, this trend bears many similarities to that observed by Labadie and Stille describing the reactivity of organostannanes (Scheme 5).20 An important difference is the inversion of the reactivity series for substituted arenes – with electron-donating groups promoting transmetalation for organosilanes, and electron-withdrawing groups promoting transmetalation with organostannanes. This is consistent with a mechanism in which sufficient activation of the silane (e.g. by added fluoride) renders Si–C bond cleavage non-rate-limiting, with instead the nucleophilicity of the silane and its capacity to displace the phosphine ligand rate-limiting.
A summary of the rates at which different organo-silicon reagents underwent transmetalation in Hiyama cross-coupling reactions. The reactivity series is based on comparisons made over several studies.44,56–59
As with transmetalation reactions involving organostannanes, very little reactivity data exist for the reaction of silanes with nickel species. An early study from Kumada’s group found that the coupling of potassium (styryl)pentafluorosilicate with iodo- or bromobenzene did not proceed in the presence of [(dppp)NiCl2], but did if the analogous Pd catalyst was used.42 In light of oxidative addition and reductive elimination typically being rapid at nickel (or at least faster than Pd),5,6,60,61 this hinted at issues with transmetalation and suggested this key step was slower at Ni than Pd. Similar indications came from Powell and Fu, who developed conditions under which secondary bromoalkanes could be coupled with trifluoro(aryl)silanes using nickel catalysis.62 Notably, only trifluoro(aryl)silanes proved effective as transmetalating agents, with the use of less reactive species such as PhSi(OMe)3 or PhSi(Me)2OH resulting in less than 10% yield.62 Similarly, Tang and co-workers reported success with distally hydroxylated silanes 26 (Scheme 11), with these serving as particularly active transmetalating agents and highlighting the difficulty typically associated with transmetalation between Si and Ni.63,64
The distally hydroxylated silanes 26 employed by Tang and co-workers to achieve cross-coupling reactivity in the presence of a nickel catalyst.64

Transmetalation with organo-boron reagents – the Suzuki–Miyaura coupling
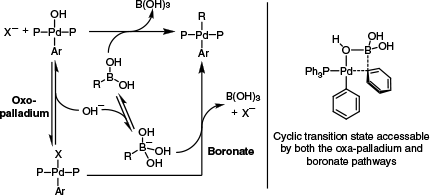
Transmetalation with organo-boron reagents also follows many of the same trends observed with stannanes and silanes. Complicating matters, there has long been discussion surrounding the role of water and inorganic bases, both of which are typically required for Suzuki–Miyaura coupling reactions.65 Two pathways have been proposed to account for this (Scheme 12): one in which the base attacks and activates the Lewis-acidic boron of the transmetalating agent (termed the boronate pathway) and another involving anion exchange at Pd (termed the oxo-palladium pathway). Experimental data from the groups of Jutand, Hartwig and Schmidt are in agreement that the oxo-palladium pathway is kinetically preferred,66–68 though the boronate pathway may occur under some circumstances and has been implicated in computational studies.69,70 Importantly, regardless of the pathway, the key Pd–C bond making step occurs by a similar transition state (Scheme 12, right), reminiscent of the cyclic SE2 pathway invoked for silanes and stannanes (Scheme 3). Such transition states have been suggested based on computational data for boron-to-palladium transmetalation,69 as well as boron-to-nickel transmetalation.71,72
(Left) The oxo-palladium and boronate pathways for transmetalation with boronic acids. (Right) The cyclic transition state accessible by either pathway as modelled by Ortuño and colleagues.69
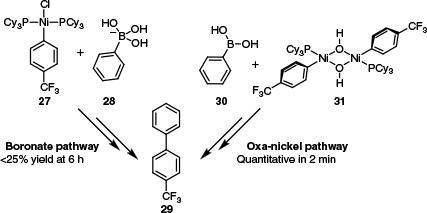
A large body of experimental data are available for boron-to-nickel transmetalation, relative to the limited data that exist for other transmetalating agents. As such, several attributes of these studies will be discussed in detail. In an important study from Monfette’s group, it was established that, like Pd, transmetalation occurred much more readily by the oxo-nickel pathway than the boronate pathway.73 This trend was established by treating [(PCy3)2Ni(Ar)Cl] 27 with PhB(OH)3K 28, and [(PCy3)Ni(Ar)(μ-OH)]2 31 with PhB(OH)2 30. The combination of [(PCy3)2Ni(Ar)Cl] 27 and PhB(OH)3K 28 resulted in very slow transmetalation, with less than 25% conversion after 6 h. By contrast, the treatment of [(PCy3)Ni(Ar)(μ-OH)]2 31 with PhB(OH)2 30 afforded quantitative conversion in 2 min (Scheme 13).
Reaction conditions and outcomes used to determine that the oxo-nickel transmetalation pathway is kinetically favoured over the boronate pathway.
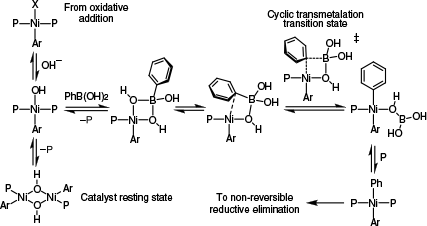
A subtle variation of the oxo-nickel mechanism suggested by the Monfette group73 was recently reported by Grimaud’s group.71 Based on extensive kinetic data, Grimaud’s group suggested that [(P)2Ni(Ar)OH] was the active species involved in transmetalation and that it existed in a complex set of equilibria in which the μ-OH species invoked by Monfette and colleagues was actually a catalyst resting state (Scheme 14).71,73 Although of great value, this study was limited to the examination of three complexes: [(PPh3)2Ni(Ar)Cl], [(PPh3)2Ni(Ar)Br] and [(PCy3)2Ni(Ar)Cl], all of which reacted quickly, as might be expected of complexes bearing mono-dentate ligands.24 From the perspective of informing rational reaction design, it is important to note that [(PPh3)2Ni(Ar)Cl] reacted more quickly than its PCy3 counterpart, suggesting an inverse relationship between transmetalation rate and ligand binding strength, as observed with stannanes and silanes.24,52,53
Insight into the rate of transmetalation has also come from reactivity trends in catalytic studies. Monteiro’s group,74 as well as that of Garg et al.,72 found the reaction rate to be dependent upon the electronic properties of the arylboronic acid, but not that of the electrophile employed, indicating that the overall rate of the catalytic reaction was dependent on the rate of transmetalation.72,74 In a related approach, rate-limiting transmetalation was suggested by Fu’s group on the basis of 1st order dependence in boronic acid, and 0th order dependence in the electrophile.75 As a notable contradiction to these studies, Miyaura’s group reported that for the nickel-catalysed coupling of chloroarenes with arylboronic acids, oxidative addition was rate-limiting and the presence of electron-donating or withdrawing groups on the boronic acid had no effect on reaction rate.76
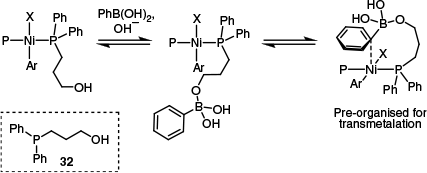
In a state-of-the-art study, Diao’s group harnessed these mechanistic insights – that transmetalation is typically rate limiting, that the oxo-nickel pathway is kinetically favoured and that μ-OH species represent a catalyst resting state – and rationally designed an exceptionally competent ligand 32.77 This involved the incorporation of a hemi-labile, distal-hydroxyl group to the phosphine in order to inhibit the formation of catalytically unproductive μ-OH dimers and permit activation of the boronic acid in preparation for subsequent intramolecular transmetalation (Scheme 15). Compared to the analogous but hydroxyl-free ligand, P(Ph)2Me, the distally hydroxylated phosphine afforded a substantial increase in reaction yield (42 v. 98% yield) and a 25-fold increase in reaction rate, demonstrating the power of mechanism-guided reaction design.
A nickel complex featuring a distally hydroxylated ligand 32 examined by Diao and co-workers.77 Prepresents the same ligand, but structural details were omitted for clarity.
As an important note, in light of the typical oxo-nickel mechanism, boron-to-nickel transmetalation generally requires displacement of the halide in [(PP)Ni(R)X] by hydroxide. This ultimately negates much of the influence of the halide in dictating reaction rate. Although this simplifies the rational design of nickel-catalysed Suzuki cross-coupling reactions, it leaves ambiguity when trying to extrapolate from these data and design reactions with other transmetalating agents, particularly those that do not require a halide displacement step.38,39
Transmetalation reactions involving nickel and organo-zinc or organo-magnesium reagents
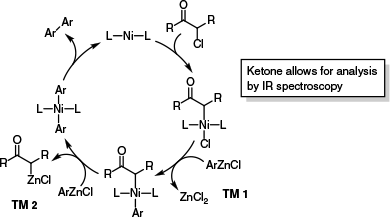
A final important study is the work of Lei’s group, where zinc-to-nickel transmetalation was examined in the context of an unusual nickel-catalysed oxidative coupling reaction.78 In a particularly elegant experiment, they avoided the challenges typically encountered when analysing Ni catalysts by NMR spectroscopy, by instead using Infrared (IR) spectroscopy and carefully selected IR-active substrates. In this study, transmetalation (Scheme 16, TM 2) was determined to be the rate-limiting step of the catalytic cycle, although this study was limited to the examination of Ni(acac)2 as the catalyst and a small number of model substrates. A similar IR spectroscopy-based strategy was recently employed by Mazet’s group to study the nickel-catalysed vinylation of phosphates with vinylmagnesium bromide, with the phosphate group serving as an IR active handle.79
Conclusion and outlook
Overall, based on the mechanistic and kinetic data available for transmetalation at palladium and nickel, several key conclusions can be made. Firstly, where data are available, similar trends appear to exist for both palladium and nickel: (1) weakly coordinating ligands generally afford faster transmetalation24,52,53,71; (2) mono-dentate ligands are generally more effective than their bidentate counterparts24,38,39,52; and (3) computational data reveal similar transition states for transmetalation at nickel and palladium.69,71,72 With this said, a general view has emerged that transmetalation at nickel is slower than at palladium, with this slow transmetalation step prohibiting the development of highly efficient nickel-catalysed cross-coupling reactions.37,42,63,64,72,74,75,77 However, these assertions are often based on limited data or are inferred from poor yields in catalysis rather than the direct measurement of the rate of transmetalation. This latter point is particularly important, as acquiring kinetic data for reactions involving nickel species is routinely frustrated by the paramagnetic nature of off-cycle catalyst species that arise.3 Looking to the future, comprehensive studies on transmetalation at a diverse range of catalytically relevant nickel complexes are needed. Determining trends associated with features such as ligand bulk, bite-angle and donicity, or how a given (pseudo)halide affects the rate of transmetalation, would enable more informed reaction design.
Data availability
Data sharing is not applicable as no new data were generated or analysed during this study.
Acknowledgements
This paper is in recognition of the 2025 Athel Beckwith Lectureship from the RACI Organic Division.
References
1 Haynes WH (Ed.). CRC Handbook of Chemistry and Physics, 95th edn. Boca Raton, FL, USA: CRC Press; 2014. doi:10.1201/b17118
2 Luescher MU, Gallou F, Lipshutz BH. The impact of earth-abundant metals as a replacement for Pd in cross coupling reactions. Chem Sci 2024; 15(24): 9016-9025.
| Crossref | Google Scholar | PubMed |
3 Ananikov VP. Nickel: the “spirited horse” of transition metal catalysis. ACS Catal 2015; 5(3): 1964-1971.
| Crossref | Google Scholar |
4 Culkin DA, Hartwig JF. Carbon–carbon bond-forming reductive elimination from arylpalladium complexes containing functionalized alkyl groups. Influence of ligand steric and electronic properties on structure, stability, and reactivity. Organometallics 2004; 23(14): 3398-3416.
| Crossref | Google Scholar |
5 Chatt J, Shaw BL. 345. Alkyls and aryls of transition metals. Part III. Nickel(II) derivatives. J Chem Soc 1960; 1960: 1718-1729.
| Crossref | Google Scholar |
6 Parshall GW. σ-Aryl compounds of nickel, palladium, and platinum. Synthesis and bonding studies. J Am Chem Soc 1974; 96(8): 2360-2366.
| Crossref | Google Scholar |
8 Fitton P, Rick EA. The addition of aryl halides to tetrakis(triphenylphosphine)palladium(0). J Organomet Chem 1971; 28(2): 287-291.
| Crossref | Google Scholar |
9 Foà M, Cassar L. Oxidative addition of aryl halides to tris(triphenylphosphine)nickel(0). J Chem Soc, Dalton Trans 1975; 1975(23): 2572-2576.
| Crossref | Google Scholar |
10 Troupel M, Rollin Y, Sibille S, Fauvarque J-F, Perichon J. Electrochemistry of nickel complexes, part 2. Reduction of triphenylphosphinenickel(II) complexes in tetrahydrofuran; reactions of zerovalent nickel with halogenobenzenes. J Chem Res 1980; 173: 24.
| Google Scholar |
11 Fauvarque J-F, Pflüger F, Troupel M. Kinetics of oxidative addition of zerovalent palladium to aromatic iodides. J Organomet Chem 1981; 208(3): 419-427.
| Crossref | Google Scholar |
12 Jutand A, Mosleh A. Rate and mechanism of oxidative addition of aryl triflates to zerovalent palladium complexes. Evidence for the formation of cationic (σ-aryl)palladium complexes. Organometallics 1995; 14(4): 1810-1817.
| Crossref | Google Scholar |
13 Casado AL, Espinet P. Mechanism of the Stille reaction. 1. The transmetalation step. Coupling of R1I and R2SnBu3 catalyzed by trans-[PdR1IL2] (R1 = C6Cl2F3; R2 = vinyl, 4-methoxyphenyl; L = AsPh3). J Am Chem Soc 1998; 120(35): 8978-8985.
| Crossref | Google Scholar |
14 Casado AL, Espinet P, Gallego AM. Mechanism of the Stille reaction. 2. Couplings of aryl triflates with vinyltributyltin. Observation of intermediates. A more comprehensive scheme. J Am Chem Soc 2000; 122(48): 11771-11782.
| Crossref | Google Scholar |
15 Nova A, Ujaque G, Maseras F, Lledós A, Espinet P. A critical analysis of the cyclic and open alternatives of the transmetalation step in the Stille cross-coupling reaction. J Am Chem Soc 2006; 128(45): 14571-14578.
| Crossref | Google Scholar | PubMed |
16 Gallego AM, Peñas-Defrutos MN, Marcos-Ayuso G, Martin-Alvarez JM, Martínez-Ilarduya JM, Espinet P. Experimental study of speciation and mechanistic implications when using chelating ligands in aryl–alkynyl Stille coupling. Dalton Trans 2020; 49(32): 11336-11345.
| Crossref | Google Scholar | PubMed |
17 Pérez-Temprano MH, Gallego AM, Casares JA, Espinet P. Stille coupling of alkynyl stannane and aryl iodide, a many-pathways reaction: the importance of isomerization. Organometallics 2011; 30(3): 611-617.
| Crossref | Google Scholar |
18 García-Melchor M, Fuentes B, Lledós A, Casares JA, Ujaque G, Espinet P. Cationic intermediates in the Pd-catalyzed Negishi coupling. Kinetic and density functional theory study of alternative transmetalation pathways in the Me–Me coupling of ZnMe2 and trans-[PdMeCl(PMePh2)2]. J Am Chem Soc 2011; 133(34): 13519-13526.
| Crossref | Google Scholar | PubMed |
19 Casado AL, Espinet P, Gallego AM, Martínez-Ilarduya JM. Snapshots of a Stille reaction. Chem Commun 2001; 2001(4): 339-340.
| Crossref | Google Scholar |
20 Labadie JW, Stille JK. Mechanisms of the palladium-catalyzed couplings of acid chlorides with organotin reagents. J Am Chem Soc 1983; 105(19): 6129-6137.
| Crossref | Google Scholar |
21 Milstein D, Stille JK. Palladium-catalyzed coupling of tetraorganotin compounds with aryl and benzyl halides. Synthetic utility and mechanism. J Am Chem Soc 1979; 101(17): 4992-4998.
| Crossref | Google Scholar |
22 Scott WJ, Stille JK. Palladium-catalyzed coupling of vinyl triflates with organostannanes. Synthetic and mechanistic studies. J Am Chem Soc 1986; 108(11): 3033-3040.
| Crossref | Google Scholar |
23 Echavarren AM, Stille JK. Palladium-catalyzed coupling of aryl triflates with organostannanes. J Am Chem Soc 1987; 109(18): 5478-5486.
| Crossref | Google Scholar |
24 Farina V, Krishnan B. Large rate accelerations in the Stille reaction with tri-2-furylphosphine and triphenylarsine as palladium ligands: mechanistic and synthetic implications. J Am Chem Soc 1991; 113(25): 9585-9595.
| Crossref | Google Scholar |
25 Napolitano E, Farina V, Persico M. The Stille reaction: a density functional theory analysis of the transmetalation and the importance of coordination expansion at tin. Organometallics 2003; 22(20): 4030-4037.
| Crossref | Google Scholar |
26 Farina V, Krishnan B, Marshall DR, Roth GP. Palladium-catalyzed coupling of arylstannanes with organic sulfonates: a comprehensive study. J Org Chem 1993; 58(20): 5434-5444.
| Crossref | Google Scholar |
27 Su W, Urgaonkar S, McLaughlin PA, Verkade JG. Highly active palladium catalysts supported by bulky proazaphosphatrane ligands for Stille cross-coupling: coupling of aryl and vinyl chlorides, room temperature coupling of aryl bromides, coupling of aryl triflates, and synthesis of sterically hindered biaryls. J Am Chem Soc 2004; 126(50): 16433-16439.
| Crossref | Google Scholar | PubMed |
28 Su W, Urgaonkar S, Verkade JG. Pd2(dba)3/P(i-BuNCH2CH2)3N-catalyzed Stille cross-coupling of aryl chlorides. Org Lett 2004; 6(9): 1421-1424.
| Crossref | Google Scholar | PubMed |
29 Naber JR, Buchwald SL. Palladium-catalyzed Stille cross-coupling reaction of aryl chlorides using a pre-milled palladium acetate and XPhos catalyst system. Adv Synth Catal 2008; 350(7–8): 957-961.
| Crossref | Google Scholar |
30 Littke AF, Fu GC. The first general method for Stille cross-couplings of aryl chlorides. Angew Chem Int Ed 1999; 38(16): 2411-2413.
| Crossref | Google Scholar | PubMed |
31 Littke AF, Schwarz L, Fu GC. Pd/P(t-Bu)3: a mild and general catalyst for Stille reactions of aryl chlorides and aryl bromides. J Am Chem Soc 2002; 124(22): 6343-6348.
| Crossref | Google Scholar | PubMed |
32 Ariafard A, Lin Z, Fairlamb IJS. Effect of the leaving ligand X on transmetalation of organostannanes (vinylSnR3) with LnPd(Ar)(X) in Stille cross-coupling reactions. A density functional theory study. Organometallics 2006; 25(24): 5788-5794.
| Crossref | Google Scholar |
33 Ye J, Bhatt RK, Falck JR. Stereospecific palladium/copper cocatalyzed cross-coupling of α-alkoxy- and α-aminostannanes with acyl chlorides. J Am Chem Soc 1994; 116(1): 1-5.
| Crossref | Google Scholar |
34 Santos LS, Rosso GB, Pilli RA, Eberlin MN. The mechanism of the Stille reaction investigated by electrospray ionization mass spectrometry. J Org Chem 2007; 72(15): 5809-5812.
| Crossref | Google Scholar | PubMed |
35 Eaborn C, Waters JA. 107. Aromatic reactivity. Part XIV. Cleavage of aryltricyclohexyl- and aryltrimethyl-stannanes by aqueous-ethanolic perchloric acid. J Chem Soc 1961; 1961: 542-547.
| Crossref | Google Scholar |
36 Cochran JC, Bayer SC, Bilbo JT, Brown MS, Colen LB, Gaspirini FJ, et al. Kinetics of the protodestannylation of vinyltrialkyltins and substituted vinyltrialkyltins. Organometallics 1982; 1(4): 586-590.
| Crossref | Google Scholar |
37 Percec V, Bae J-Y, Hill DH. Aryl mesylates in metal catalyzed homo- and cross-coupling reactions. 4. Scope and limitations of aryl mesylates in nickel catalyzed cross-coupling reactions. J Org Chem 1995; 60(21): 6895-6903.
| Crossref | Google Scholar |
38 Shirakawa E, Yamasaki K, Hiyama T. Nickel-catalysed cross-coupling reactions of aryl halides with organostannanes. J Chem Soc, Perkin Trans 1 1997; 1997(17): 2449-2450.
| Crossref | Google Scholar |
39 Shirakawa E, Yamasaki K, Hiyama T. Cross-coupling reaction of organostannanes with aryl halides catalyzed by nickel–triphenylphosphine or nickel–lithium halide complex. Synthesis 1998; 1998(10): 1544-1549.
| Crossref | Google Scholar |
40 Russell JEA, Entz ED, Joyce IM, Neufeldt SR. Nickel-catalyzed stille cross coupling of C–O electrophiles. ACS Catal 2019; 9(4): 3304-3310.
| Crossref | Google Scholar | PubMed |
41 Yamamoto K, Shinohara K, Ohuchi T, Kumada M. The reaction of vinylsilanes with dichlorobis(benzonitrile)palladium(II). Tetrahedron Lett 1974; 15(13): 1153-1156.
| Crossref | Google Scholar |
42 Yoshida J, Tamao K, Yamamoto H, Kakui T, Uchida T, Kumada M. Organofluorosilicates in organic synthesis. 14. Carbon–carbon bond formation promoted by palladium salts. Organometallics 1982; 1(3): 542-549.
| Crossref | Google Scholar |
43 Tamao K, Yoshida J, Yamamoto H, Kakui T, Matsumoto H, Takahashi M, et al. Organofluorosilicates in organic synthesis. 12. Preparation of organopentafluorosilicates and their cleavage reactions by halogens and N-bromosuccinimide. Synthetic and mechanistic aspects. Organometallics 1982; 1(2): 355-368.
| Crossref | Google Scholar |
44 Hatanaka Y, Hiyama T. Cross-coupling of organosilanes with organic halides mediated by a palladium catalyst and tris(diethylamino)sulfonium difluorotrimethylsilicate. J Org Chem 1988; 53(4): 918-920.
| Crossref | Google Scholar |
45 Hatanaka Y, Hiyama T. Palladium-mediated silylation of organic halides with disilane/F– reagent. Tetrahedron Lett 1987; 28(40): 4715-4718.
| Crossref | Google Scholar |
46 Jacobs E, Keaveney ST. Experimental and computational studies towards chemoselective C–F over C–Cl functionalisation: reversible oxidative addition is the key. ChemCatChem 2021; 13(2): 637-645.
| Crossref | Google Scholar |
47 Keaveney ST, Schoenebeck F. Palladium‐catalyzed decarbonylative trifluoromethylation of acid fluorides. Angew Chem Int Ed 2018; 57(15): 4073-4077.
| Crossref | Google Scholar | PubMed |
48 Malapit CA, Bour JR, Brigham CE, Sanford MS. Base-free nickel-catalysed decarbonylative Suzuki–Miyaura coupling of acid fluorides. Nature 2018; 563(7729): 100-104.
| Crossref | Google Scholar | PubMed |
49 Saijo H, Sakaguchi H, Ohashi M, Ogoshi S. Base-free Hiyama coupling reaction via a group 10 metal fluoride intermediate generated by C–F bond activation. Organometallics 2014; 33(14): 3669-3672.
| Crossref | Google Scholar |
50 Hatanaka Y, Hiyama T. Stereochemistry of the cross-coupling reaction of chiral alkylsilanes with aryl triflates: a novel approach to optically active compounds. J Am Chem Soc 1990; 112(21): 7793-7794.
| Crossref | Google Scholar |
51 Sugiyama A, Ohnishi Y-y, Nakaoka M, Nakao Y, Sato H, Sakaki S, Nakao Y, Hiyama T. Why does fluoride anion accelerate transmetalation between vinylsilane and palladium(II)–vinyl complex? Theoretical study. J Am Chem Soc 2008; 130(39): 12975-12985.
| Crossref | Google Scholar | PubMed |
52 Tymonko SA, Smith RC, Ambrosi A, Ober MH, Wang H, Denmark SE. Mechanistic significance of the Si–O–Pd bond in the palladium-catalyzed cross-coupling reactions of arylsilanolates. J Am Chem Soc 2015; 137(19): 6200-6218.
| Crossref | Google Scholar | PubMed |
53 Mateo C, Fernández-Rivas C, Cárdenas DJ, Echavarren AM. Intramolecular transmetalation of arylpalladium(II) and arylplatinum(II) complexes with silanes and stannanes. Organometallics 1998; 17(17): 3661-3669.
| Crossref | Google Scholar |
54 Ju J, Nam H, Jung HM, Lee S. Palladium-catalyzed cross-coupling of trimethoxysilylbenzene with aryl bromides and chlorides using phosphite ligands. Tetrahedron Lett 2006; 47(49): 8673-8678.
| Crossref | Google Scholar |
55 Denmark SE, Wu Z. Synthesis of unsymmetrical biaryls from arylsilacyclobutanes. Org Lett 1999; 1(9): 1495-1498.
| Crossref | Google Scholar |
56 Hatanaka Y, Goda K, Okahara Y, Hiyama T. Highly selective cross-coupling reactions of aryl(halo)silanes with aryl halides: a general and practical route to functionalized biaryls. Tetrahedron 1994; 50(28): 8301-8316.
| Crossref | Google Scholar |
57 Nakao Y, Oda T, K. Sahoo A, Hiyama T. Triallyl(aryl)silanes serve as a convenient agent for silicon-based cross-coupling reaction of aryl halides. J Organomet Chem 2003; 687(2): 570-573.
| Crossref | Google Scholar |
58 Li L, Navasero N. All-carbon-substituted vinylsilane stable to TBAF: synthesis of allyldimethylvinylsilane and its Pd-catalyzed cross-coupling under mild conditions. Org Lett 2006; 8(17): 3733-3736.
| Crossref | Google Scholar | PubMed |
59 Rossi R, Carpita A, Messeri T. Palladium-mediated reaction between aryl iodides and stereodefined 2-arylethenyldimethylphenylsilanes or 2-arylethynyldimethylphenylsilanes, in the presence of a fluoride-ion source. Gazz Chim Ital 1992; 122(2): 65-72.
| Crossref | Google Scholar |
60 Otsuka S, Nakamura A, Yoshida T, Naruto M, Ataka K. Chemistry of alkoxycarbonyl, acyl, and alkyl compounds of nickel(II) and palladium(II). J Am Chem Soc 1973; 95(10): 3180-3188.
| Crossref | Google Scholar |
61 Cooper AK, Burton PM, Nelson DJ. Nickel versus palladium in cross-coupling catalysis: on the role of substrate coordination to zerovalent metal complexes. Synthesis 2020; 52(4): 565-573.
| Crossref | Google Scholar |
62 Powell DA, Fu GC. Nickel-catalyzed cross-couplings of organosilicon reagents with unactivated secondary alkyl bromides. J Am Chem Soc 2004; 126(25): 7788-7789.
| Crossref | Google Scholar | PubMed |
63 Tang S, Li S-H, Nakao Y, Hiyama T. Aryl[2-(hydroxypro-2-yl)cyclohyxyl]dimethylsilane: a robust aryl(trialkyl)silane reagent for nickel-catalyzed cross-coupling reactions with aryl tosylates. Asian J Org Chem 2013; 2(5): 416-421.
| Crossref | Google Scholar |
64 Tang S, Takeda M, Nakao Y, Hiyama T. Nickel-catalysed cross-coupling reaction of aryl(trialkyl)silanes with aryl chlorides and tosylates. Chem Commun 2011; 47(1): 307-309.
| Crossref | Google Scholar | PubMed |
65 Lennox AJJ, Lloyd-Jones GC. Transmetalation in the Suzuki–Miyaura coupling: the fork in the trail. Angew Chem Int Ed 2013; 52(29): 7362-7370.
| Crossref | Google Scholar | PubMed |
66 Amatore C, Jutand A, Le Duc G. Kinetic data for the transmetalation/reductive elimination in palladium-catalyzed Suzuki–Miyaura reactions: unexpected triple role of hydroxide ions used as base. Chem Eur J 2011; 17(8): 2492-2503.
| Crossref | Google Scholar | PubMed |
67 Carrow BP, Hartwig JF. Distinguishing between pathways for transmetalation in Suzuki–Miyaura reactions. J Am Chem Soc 2011; 133(7): 2116-2119.
| Crossref | Google Scholar | PubMed |
68 Schmidt AF, Kurokhtina AA, Larina EV. Role of a base in Suzuki–Miyaura reaction. Russ J Gen Chem 2011; 81(7): 1573-1574.
| Crossref | Google Scholar |
69 Ortuño MA, Lledós A, Maseras F, Ujaque G. The transmetalation process in Suzuki–Miyaura reactions: calculations indicate lower barrier via boronate intermediate. ChemCatChem 2014; 6(11): 3132-3138.
| Crossref | Google Scholar |
70 D’Alterio MC, Casals-Cruañas È, Tzouras NV, Talarico G, Nolan SP, Poater A. Mechanistic aspects of the palladium-catalyzed Suzuki–Miyaura cross-coupling reaction. Chem Eur J 2021; 27(54): 13481-13493.
| Crossref | Google Scholar | PubMed |
71 Payard P-A, Perego LA, Ciofini I, Grimaud L. Taming nickel-catalyzed Suzuki–Miyaura coupling: a mechanistic focus on boron-to-nickel transmetalation. ACS Catal 2018; 8(6): 4812-4823.
| Crossref | Google Scholar |
72 Quasdorf KW, Antoft-Finch A, Liu P, Silberstein AL, Komaromi A, Blackburn T, Ramgren SD, Houk KN, Snieckus V, Garg NK. Suzuki–Miyaura cross-coupling of aryl carbamates and sulfamates: experimental and computational studies. J Am Chem Soc 2011; 133(16): 6352-6363.
| Crossref | Google Scholar | PubMed |
73 Christian AH, Müller P, Monfette S. Nickel hydroxo complexes as intermediates in nickel-catalyzed Suzuki–Miyaura cross-coupling. Organometallics 2014; 33(9): 2134-2137.
| Crossref | Google Scholar |
74 Zim D, Lando VR, Dupont J, Monteiro AL. NiCl2(PCy3)2: a simple and efficient catalyst precursor for the Suzuki cross-coupling of aryl tosylates and arylboronic acids. Org Lett 2001; 3(19): 3049-3051.
| Crossref | Google Scholar | PubMed |
75 Lu Z, Wilsily A, Fu GC. Stereoconvergent amine-directed alkyl–alkyl Suzuki reactions of unactivated secondary alkyl chlorides. J Am Chem Soc 2011; 133(21): 8154-8157.
| Crossref | Google Scholar | PubMed |
76 Saito S, Oh-tani S, Miyaura N. Synthesis of biaryls via a nickel(0)-catalyzed cross-coupling reaction of chloroarenes with arylboronic acids. J Org Chem 1997; 62(23): 8024-8030.
| Crossref | Google Scholar | PubMed |
77 Yang J, Neary MC, Diao T. ProPhos: a ligand for promoting nickel-catalyzed Suzuki–Miyaura coupling inspired by mechanistic insights into transmetalation. J Am Chem Soc 2024; 146(9): 6360-6368.
| Crossref | Google Scholar | PubMed |
78 Jin L, Xin J, Huang Z, He J, Lei A. Transmetalation is the rate-limiting step: quantitative kinetic investigation of nickel-catalyzed oxidative coupling of arylzinc reagents. J Am Chem Soc 2010; 132(28): 9607-9609.
| Crossref | Google Scholar | PubMed |
79 Poisson P-A, Tran G, Besnard C, Mazet C. Nickel-catalyzed Kumada vinylation of enol phosphates: a comparative mechanistic study. ACS Catal 2021; 11(24): 15041-15050.
| Crossref | Google Scholar |
Dr Nicholas Solomon received his PhD in 2025 from Macquarie University in Sydney, Australia, having studied organometallic reaction mechanisms under the supervision of Dr Sinead Keaveney. Following this, he joined the Dementia Research Centre at Macquarie University as a post-doctoral researcher in the group of Ole Tietz. His work now focuses on developing small molecule and peptide-based therapeutics, as well as synthetic strategies towards these applications. |
Dr Sinead Keaveney received her PhD in 2016 from UNSW Australia, in the field of physical organic chemistry under the supervision of Assoc. Prof. Jason Harper. Following a post-doctoral position at RWTH Aachen University, Germany, with Prof. Franziska Schoenebeck, and a research fellowship at Macquarie University, Australia, she joined the University of Wollongong in 2021 as a lecturer. Her research program is focused on developing new synthetic methodology through combined experimental, computational and mechanistic studies. |