

Liver microsome stability of N-methylated cyclic hexapeptides is decreased by the presence of cis-amide bonds
Huy N. Hoang A B , David P. Fairlie A B * and Timothy A. Hill A B *
A B *
A
B
Abstract
N-methylation of cyclic peptides is a widely used strategy to enhance membrane permeability; however, it can also influence metabolic stability. In celebration of Professor David Craik’s scientific achievements – particularly in the field of peptide research – we were fortunate to gain access to a series of his cyclic peptides to investigate their liver microsomal stability. Our study revealed that the liver microsome stability of a series of 14 cyclic hexapeptides is highly variable, despite minimal differences in sequence, molecular weight and cLogP. Notably, all compounds containing cis-amide bonds exhibited very poor rat liver microsomal stability, with half-lives of less than 3 min. This work highlights a potential metabolic liability that should be taken into account when designing cyclic peptides as potential drug candidates.
Keywords: cis-amide bonds, cyclic peptides, drug clearance, hepatic metabolism, metabolic stability, n-methylation, oral bioavailability, peptide structure.
Introduction
Low cell permeability and limited oral bioavailability restrict the therapeutic application of peptides.1–4 Cyclisation and N-methylation have been widely used to enhance gastric stability and improve membrane permeability; however, limited attention has been given to how these modifications may affect hepatic stability.5–7
Hepatic stability is a key factor, along with plasma binding and gastric stability, in ensuring the survival of orally delivered drugs through first-pass metabolism before reaching systemic exposure.8,9 Prediction of hepatic stability in vivo is often based on in vitro assays using hepatocytes, liver microsomes or enzyme assays.10,11 In the development of orally bioavailable cyclic peptides, liver microsome stability is commonly used either to select potentially suitable compounds for further pharmacokinetic studies or to help explain the clearance rates observed when compounds are administered orally.12,13 Limited liver microsome stability data on cyclic peptides suggest that stability can be significantly influenced by the introduction of N-methyl groups, often leading to poor oral bioavailability despite high membrane permeability.12,14 The structural features responsible for this loss of stability in some cyclic peptides have not been widely explored.
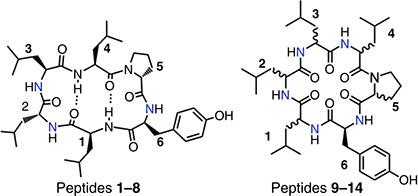
The cyclic hexapeptide scaffold [Leu-Leu-Leu-Leu-Pro-Tyr] has been widely exploited to develop peptides with high cell and membrane permeability by altering peptide stereochemistry and N-methylation pattern.15–21 Some of these peptides were also shown to have oral bioavailability.5,17 In 2015, the Craik group in, collaboration with Pfizer, published a library of 62 cyclic hexapeptides and reported on the features and properties that drive Caco 2 cell and parallel artifical membrane permeability (PAMPA).18 Here, we extend this study by examining the rat liver microsome stability of 14 of these compounds that have the [Leu-Leu-Leu-Leu-Pro-Tyr] base scaffold (Table 1) and displayed Caco-2 cell monolayer permeability.18
 | |||||||
|---|---|---|---|---|---|---|---|
| Cyclic peptide | Residue position | ||||||
| 1 | 2 | 3 | 4 | 5 | 6 | ||
| Chirality and N-methylation | |||||||
| 1 | L | D | L | L | D | L | |
| 2 | L | D | L | L | D | L | |
| 3 | L | D | L | L | D | L | |
| 4 | L | D | L | L | D | L | |
| 5 | L | D | L | L | D | L | |
| 6 | L | D | L | L | D | L | |
| 7 | L | D | L | L | D | L | |
| 8 | L | D | L | L | D | L | |
| 9 | L | D | D | D | L | L | |
| 10 | D | D | D | L | L | L | |
| 11 | D | D | L | D | L | L | |
| 12 | L | D | L | L | D | L | |
| 13 | D | D | L | D | L | L | |
| 14 | D | L | L | L | L | L | |
Here, we report striking differences in intrinsic clearance rates of these 14 compounds despite limited variations in molecular weight, cLogP and stereochemistry. Significantly, compounds with poor rat liver microsomal stability consistently feature conformers containing a cis-amide bond. These findings highlight the importance of avoiding this structural motif in the design of cyclic peptides with good drug-like properties.
Results and discussion
Within the library of 14 cyclic peptides screened, a range of microsomal stabilities was observed (Table 1, Fig. 1) despite all compounds containing the same sequence of amino acids, only varying in stereochemistry and N-methylation pattern. Liver microsomal processing of the compounds was NADPH dependent (Supplementary Fig. S1), as minimal processing was observed after 1 h in its absence. This also indicated that the compounds did not display high rat liver microsomal protein binding. The most unstable compounds 9–13 all contained two adjacent N-methyl amino acids in positions 1–2; apart from this, no other discernible trend is observed to account for a compound's stability or lack thereof. To highlight this, the intrinsic clearance was plotted against the number of N-methyl groups (Fig. 2a) and the number of D-amino acids (Fig. 2b), as these have been reported as possible contributors to metabolic instability,22,23 as well as against measured properties related to compound hydrophobicity LogP (Fig. 2c) and Caco-2 cell monolayer permeabilities (Fig. 2d). In all cases, no clear trend emerges that could reliably predict microsomal stability.
Rat liver microsomal stability expressed as intrinsic clearance (CLint) of compounds 1–14, with stable compounds green, unstable compounds orange and highly unstable compounds red.
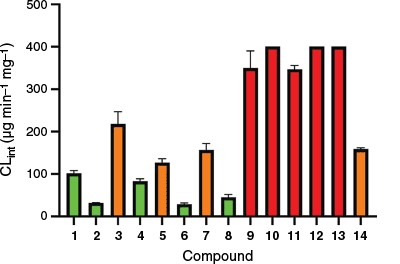
Rat liver microsome clearance v. (a) number of NMe, (b) number of D-amino acids, (c) LogP or (d) Caco-2 cell mono-layer permeability.18
For compounds with one N-methyl group (2, 3, 4), different intrinsic clearance rates were observed (Fig. 2a), with compound 3 having the fastest enzymatic processing, showing intrinsic clearance at 200 µg mL−1 min−1. For compounds with two N-methyl groups or three N-methyl groups, compounds were distributed across all three stability classes. The lone compound with four N-methyl groups (12) is highly unstable, but no conclusions can be drawn due to the lack of comparators. The position of N-methylation relative to both the amide number and the proximity to other methylations appears to influence microsomal stability (Table 1). All five of the most unstable compounds (9–13) have N-methylation at both the 1 and 2 amide positions, whereas compound 14, with moderate stability, is the only other compound containing this motif. Adjacent N-methylated amino acids, as observed in compounds 5 and 8, are tolerated; notably, all three stable compounds with this motif (14, 5 and 8) feature adjacent amino acids with D,L stereochemistry. By contrast, the unstable compounds (9–13) exhibit L,D or D,D stereochemistry. N-methylation of amides 1 and 2 simultaneously appears to introduce a metabolic liability into the cyclic peptide. The logP (Fig. 2c) of the compounds also does not appear to drive instability, as the most unstable compounds have logP between 4.5 and 5.5. Plotting intrinsic clearance against reported Caco-2 permeability (Fig. 2d) reveals that compounds can exhibit high permeability while displaying a wide range of liver microsome stabilities, as observed for compounds 8, 10, 11 and 14. Since neither the bulk chemical properties nor the experimental hydrophobicity-related measures could explain the differences in microsomal susceptibility, we turned to examining the three-dimensional (3-D) structures of the compound library.
We and others have previously reported the 3-D solution structures of peptides 1, 2, 3, 4 and 8.5,19,20 These peptides all share a common, backbone-stabilised conformation in solution, forming a β-hairpin shape characterised by two type II′ β-turns at each end. The structure is stabilised by two intramolecular hydrogen bonds and a motif involving D- and L-amino acid residues. As compounds 1–8 all contain the same stereochemical conformation and all are either highly stable (Fig. 1, green) or moderately stable (Fig. 1, orange) to rat liver microsomal metabolism, it appears that this cyclic backbone structure does not present any major metabolic liabilities.
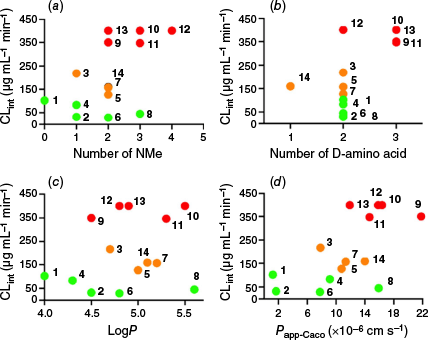
Possible 3-D structures of peptides 9–14 were explored using a conformational search in MacroModel modeule (part of the Schrodinger Suite, ver. 2024-3, see https://www.schrodinger.com/platform/products/macromodel/) with the OPLS4 force field (Fig. 3). Unlike peptides 1, 2, 3, 4 and 8, which adopted a common rectangular shape, peptides 9–14 are predicted to favour asymmetric conformations, featuring different turns at each end and containing either one or no intramolecular hydrogen bonds. The predicted structures of peptides 9–13 all contain a cis-amide bond involving one of the N-methylated amides (Fig. 3), whereas peptide 14 exhibited exclusively trans-amide bonds. In the cis configuration, the electronegative carbonyl oxygen and the N-methyl group are positioned on the same side of the amide bond.
Computer predicted structures for 9–14. Predicted 3-D structures generated using the ConfGen module in the Schrödinger Suite (ver. 2024-3, see https://www.schrodinger.com/platform/products/confgen/), highlighting the location of the single cis-amide bond (at position 1 compounds 9, 11 and 12; at position 2 compounds 10 and 13) but none in 14 and intramolecular H-bonds in dashed lines.
Computer modelling predicted that compounds 9–13 adopt a cis-amide conformation. In light of this, we re-examined the raw NMR data from the original publication18 for all 14 compounds. Our analysis revealed that, in certain solvent systems, compounds 9–13 exhibited multiple conformations in solution. Since all original spectra were recorded with water suppression, we performed extended ROE experiments to specifically investigate strong ROE interactions between αi-and-αi+1 protons. (Fig. 4a). All compounds with high intrinsic clearances (9, 10, 11, 12 and 13) were found to contain a cis-amide bond in DMSO-d6 or CDCl3, whereas all other compounds did not (Fig. 4b). The data suggest that within this series, the presence of a cis-amide bond introduces a metabolic liability.
(a) The trans and cis configurations of an amide bond exhibit distinct distances and ROE patterns between the α-protons of two consecutive residues. (b) The relative populations of the cis isomer in different solvents – DMSO-d6 (black) and CDCl3 (grey) – were quantified using ROESY experiments. ROE intensities between consecutive α-protons were measured for compounds 1–14, where medium to strong ROE signals indicate the presence of a cis-amide bond between the corresponding residues. (c) NMR-derived structures of 13 in CDCl3: Superimposition of the 10 lowest-energy conformers. An intramolecular hydrogen bond is indicated by a dashed line. Backbone carbons are shown in yellow, sidechain carbons in green, nitrogen atoms in blue and oxygen atoms in red.
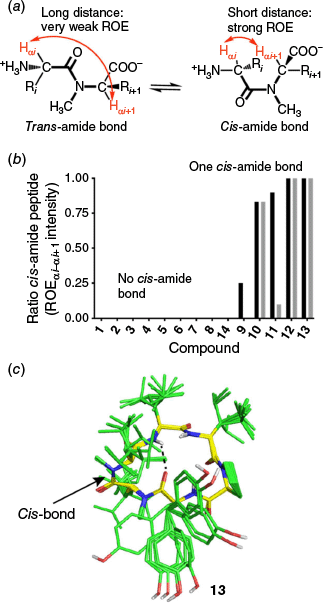
The cis-amide bond does not appear to be restricted to one position. Compounds 9 and 12 exhibit the cis conformation at position 2, whereas compounds 10, 11 and 13 have it at position 1 (Fig. 3). The stereochemistry of the amino acid with the N-methylation also does not appear to affect stability, as both D and L configurations are involved, with L amino acids accounting for four of the five unstable compounds. These results suggest that the cis-amide bond itself is the cause of microsomal instability, as no single clear motif is found across all unstable compounds.
To validate our structural predictions, the NMR structure of compound 13 was determined in CDCl3, confirming the presence of a cis-amide bond (Fig. 4c). An overlay of the model and the lowest-energy NMR structure showed good agreement, with a Cα RMSD of 0.75 Å based on alignment in PyMOL (ver. 3.1.1, see https://www.pymol.org/).
As the presence of a cis-amide bond may affect the hydrophobic surface of the cyclic peptide, we calculated the area of the largest connected hydrophobic surface patches of 1–14 using the Protein Surface Analysis module in the Schrödinger Suite (ver. 2024-3) with the OPLS4 force field. We have previously shown that maximising larger hydrophobic surface area patches can be a key driver of cyclic peptide permeability, an effect we reported for compounds 1–4.20 However, the impact of this on microsome stability has yet to be explored.
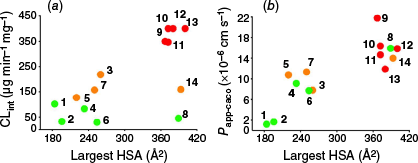
Plotting the calculated largest hydrophobic surface patch area against rat liver microsomal stability (Fig. 5a) reveals that, in general, compounds with larger hydrophobic surface patch areas, such as 9–13, exhibit poor stability. However, compounds 8 and 14, despite having similarly large hydrophobic surface patch areas, show greater stability, indicating that a large hydrophobic surface alone does not necessarily drive instability. This further highlights the signifigance of the role of cis-amide bonds in promoting rat liver microsomal instability. By contrast, plotting the same largest hydrophobic surface area data against previously reported permeability values (Fig. 5b) confirms it as a strong predictor of permeability within this compound series.
(a) Area of the largest hydrophobic surface patch v. microsomal stability CLint. (b) Area of the largest hydrophobic surface patch v. Caco-2 cell monolayer permeability.18
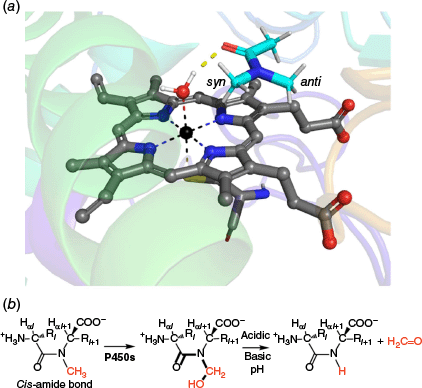
To investigate why the presence of a cis-amide bond in a cyclic peptide may lead to microsomal instability, we performed molecular docking of N,N-dimethylacetamide (DMA) into the heme active site of human cytochrome P450 3A4 using the crystal structure (PDB code: 1W0F) to investigate its binding geometry (Fig. 6). The results showed that the carbonyl oxygen of DMA forms a hydrogen bond with a heme-bound water molecule coordinated to the iron centre. Notably, the syn N-methyl group is oriented toward the heme-bound water, whereas the anti N-methyl group is projected away from it. A similar orientation has been observed in the interactions of both natural and synthetic compounds with cytochrome P450 enzymes.24 This binding configuration suggests that the amide oxygen, in a cis-amide conformation, plays a key role in directing the cis N-methyl group toward the activated water, thereby positioning it for potential hydroxylation by the enzyme.
(a) Top-ranked docking pose of N,N-dimethylacetamide bound to the heme active site of CYP3A4 (PDB: 1W0F). (b) Proposed mechanism for P450-mediated hydroxylation of a cis-amide bond.
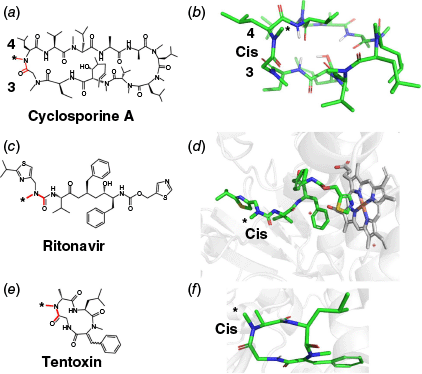
Several examples of N-demethylation of N-methyl amide–containing compounds by cytochrome P450 enzymes have been reported.25–30 Our analysis reveals that in nearly all cases, the N-methyl group undergoing demethylation is syn to the carbonyl oxygen atom. Notably, cyclosporine A (CSA) undergoes P450-mediated metabolism resulting in N-demethylation between residues 3 and 4 (Fig. 7a, b).25,26 This amide bond has been observed in both cis and trans configurations depending on the environment (Fig. 7b).25 CSA, in total, contains four N-methylated amide bonds. Similarly, one of the major metabolites of ritonavir is formed by N-demethylation (Fig. 7c). The corresponding amide bond is also reported to exist in both cis and trans forms, but adopts a cis configuration when bound to CYP3A4 (Fig. 7d).27–29 Another example is the cyclic peptide tentoxin,30 which contains two cis-amide bonds (Fig. 7e, f). However, only the N-methyl group on the alanine residue undergoes hydroxylation by cytochrome P450, whereas the other N-methyl group is sterically shielded by bulky side chains (isobutyl and phenyl). These data support the findings presented in this study. They underscore the importance of amide bond geometry, especially the cis configuration, in determining the lability of N-methyl groups, which has significant implications for microsomal instability and ultimately the oral bioavailability of drugs.
(a, b) Structures of cyclosporine A, highlighting the cis-amide bond between residues 9 and 10 (CCCD: DEKSAN). (c) 2-D structure for ritonavir and (d) 3-D structure of ritonavir bound to CYP 3A4 (PDB: 5VCE). (e) 2-D structure of tentoxin; (f) 3-D structure of tentoxin bound to spinach chloroplast F1-ATPase (PDB: 1KMH). Cis-amide bonds are highlighted in red or labelled as ‘cis’; active N-methyl groups are marked with an asterisk (*).
Conclusion
N-methylation is a widely used strategy to improve the membrane permeability of cyclic peptides, sometimes leading to compounds with promising oral bioavailability.5–7 However, in many studies, hepatic or microsomal stability is considered a secondary concern, and there remains limited guidance in the literature for designing cyclic peptides with both high permeability and acceptable first-pass metabolic stability. In this study, we demonstrate that within a library of N-methylated cyclic hexapeptides, microsomal stability varies markedly, even among compounds with comparable Lipinski-like parameters. Structural analysis by modelling and NMR revealed that peptides with adjacent N-methyl groups at positions 1 and 2 often exhibit poor stability, coinciding with the presence of a cis-amide bond at one of those positions. Our findings suggest that the cis-amide conformation, rather than N-methylation alone, is a key contributor to metabolic instability. These results highlight an important trade-off; whereas N-methylation can enhance cellular permeability by increasing hydrophobicty it may also introduce a cis-amide, thereby introducing a metabolic liability that limits the compound’s oral bioavailability. This underscores the need to balance conformational and metabolic considerations when optimising cyclic peptide scaffolds for drug development.
Experimental
Rat liver microsome stability
Rat liver microsomes were purchased from Sigma–Aldrich. Microsomes stored at −80°C were defrosted on crushed ice. Test compounds (185 µL, 1 µM in 100 mM of phosphate buffer pH 7.4, containing 0.1% DMSO) were added to microsomes (5 µL) in 1-mL Eppendorf tubes. These were heated in a circulating water bath at 37°C for 5 min before cofactor NADPH (10 µL, 20 mM) was added. Aliquots (20 µL) were taken at time points 0, 5, 15, 30 and 60 min from the addition of NADPH. Aliquots were added to MeCN containing 1 μM of caffeine as internal standard (100 µL) and centrifuged at 13,000g for 3 min at 25°C. Supernatants were analysed by LC-MS or LC-MSMS for degradation of substrate. Concentrations were plotted as a function of time in a semi-logarithmic plot. Intrinsic clearance rates (CLint) were calculated from the formula (Eqn 1):
where V is the volume of solution used (µL mg–1), volume of incubation (µL) and protein in the incubation (mg), and
Experimental logP
logP measurements were conducted as reported in Hoang et al.20 UPLC analysis was performed by measuring light absorption at wavelength 214 nm on a Shimadzu UHPLC system (LC-30AD, SIL-30AC, CBM-20A, SPD-M20A, CTO-20A) using solvent mixtures of 0.1% trifluoroacetic acid in water (buffer A) and 0.1% trifluoroacetic acid in acetonitrile (MeCN)/H2O (9:1) (buffer B) with a flow rate of 0.5 mL min−1 on an Eclipse Plus C8 column (2.1 µm × 100 mm). Retention times of standards with reported logP retention times were measured. logP values were used from the OECD Guidelines for the testing of chemicals.31 Retention times of 1–14 were measured under the same conditions. logP was calculated from the standard curve.
NMR structure calculations
1-D and 2-D 1H-NMR spectra were recorded on a 600-MHz Bruker Avance III DRX-600 spectrometer with cryoprobe. 2-D 1H-NMR spectra were recorded in phase-sensitive mode using time-proportional phase incrementation for quadrature detection in the t1 dimension. The 2-D experiments recorded were TOCSY (standard Bruker mlevgpph pulse program), ROESY (standard Bruker roesygpph pulse program) and dqfCOSY (standard Bruker dqfcosygpph pulse program). TOCSY spectra were acquired over 6887 Hz with 2048 complex data points in F2, 256 increments in F1 and 8 scans per increment. ROESY spectra were acquired over 6887 Hz with 4096 complex data points in F2, 512 increments in F1 and 32 scans per increment. TOCSY and ROESY spectra were acquired with several isotropic mixing times of 80 ms for TOCSY, 250–350 ms for ROESY. The variable temperature NMR experiments were performed in 10°C increments over the range of 288–318 K. Spectra were processed using TopSpin (ver. 4.5.0, Bruker, Germany, see https://www.bruker.com/en/products-and-solutions/mr/nmr-software/topspin.html). The t1 dimensions of all 2-D spectra were zero-filled to 1024 real data points with 90° phase-shifted QSINE bell window functions applied in both dimensions, followed by Fourier transformation and fifth order polynomial baseline correction. 1H chemical shifts were referenced to DMSO (δ 2.50 ppm). 3JNHCHα coupling constants were measured from 1-D 1H NMR and dqf-COSY spectra.
The distance restraints used for calculating NMR solution structures in CDCl3 were derived from ROESY spectra (recorded at 298 K) using a mixing time of 350 ms. ROE cross-peak volumes were classified manually as strong (upper distance constraint ≤ 2.7 Å), medium (≤3.5 Å), weak (≤5.0 Å) and very weak (≤6.0 Å). Standard pseudoatom distance corrections were applied for non-stereospecifically assigned protons. To address the possibility of conformational averaging, intensities were classified conservatively and only upper distance limits were included in the calculations to allow the largest possible number of conformers to fit the experimental data. Backbone dihedral angle restraints were inferred from 3JNHCHα coupling constants in 1-D spectra, ϕ was restrained to –65 ± 30° for 3JNHCHα ≤6 Hz and to −120 ± 30° for 3JNHCHα ≥8 Hz. The cis/trans-amide conformations about peptide bonds were determined with or without the presence of strong CHα–CHα (i, i + 1) ROEs respectively in the ROESY spectra, so the ψ-angles could be set to cis (ψ = 0°) and trans (ψ = 180°) accordingly. Starting structures with randomised ϕ and ψ angles and extended side chains were generated using an ab initio simulated annealing protocol. The calculations were performed using the standard force field parameter set (PARALLHDG5.2.PRO) and topology file (TOPALLHDG5.2.PRO) in XPLOR-NIH (ver. 3.10, see https://nmr.cit.nih.gov/xplor-nih/) with in-house modifications to generate end-to-end cyclisation. Refinement of structures was achieved using the conjugate gradient Powell algorithm with 4000 cycles of energy minimisation and a refined force field based on the program CHARMm (ver. 36, see https://academiccharmm.org/program/). Structures were visualised with PyMOL and analysed for distance (>0.2 Å) and dihedral angle (>5°) violations using noe.inp files in XPLOR-NIH. Final structures contained no distance violations (>0.2 Å) or angle violations (>5°).
Conformational search
Three-dimensional structures of peptides 8–13 were generated using the Conformational Search module in MacroModel, part of the Schrödinger Suite. Initial structures were built using the Builder module and processed through the Protein Preparation Wizard (ver. 2024-3, see https://www.schrodinger.com/platform/products/maestro/) to assign protonation states at pH 7 ± 2. Conformational searches were performed using the OPLS4 force field following energy minimisation and structural refinement. Representative low-energy conformers were manually selected and visualised using PyMOL.
Molecular docking
The crystal structure of human cytochrome P450 3A4 (PDB ID: 1W0F) was prepared for molecular docking using the Protein Preparation Wizard in the Schrödinger Suite (ver. 2024-2). Protonation states of ionisable residues were assigned for pH 7.4 ± 1.0 using Epik (see https://www.schrodinger.com/platform/products/epik/), and the N- and C-termini were capped with neutral acetyl and N-methylamide groups respectively. All crystallographic water molecules were removed prior to docking. The protein structure was subsequently energy-minimised using the OPLS4 force field. Ligand structures were prepared with LigPrep (ver. 2024-2, see https://www.schrodinger.com/platform/products/ligprep/), with protonation states also set for pH 7.4 ± 1.0 using Epik, followed by energy minimisation using the OPLS4 force field. Molecular docking was performed using Glide in Standard Precision (SP) mode. The receptor grid was centred on the co-crystallised ligand and encompassed residues within a 10-Å cube. Docking poses with the highest predicted binding affinities were selected for further analysis.
Data availability
The data that support this study will be shared upon reasonable request to the corresponding author.
Declaration of funding
We acknowledge the Australian Research Council Centre of Excellence for Innovations in Peptide and Protein Science grant CE200100012 (to D. P. Fairlie) and the National Health and Medical Research Council of Australia Leadership Fellowship (2009551) (to D. P. Fairlie) for funding this research.
Acknowledgements
We thank David Craik for providing compounds 1–14 used for experiments in this manuscript.
References
1 Muttenthaler M, King GF, Adams DJ, Alewood PF. Trends in peptide drug discovery. Nat Rev Drug Discov 2021; 20: 309-325.
| Crossref | Google Scholar | PubMed |
2 Craik DJ, Fairlie DP, Liras S, Price D. The future of peptide-based drugs. Chem Biol Drug Des 2013; 81: 136-147.
| Crossref | Google Scholar | PubMed |
3 Nielsen DS, Shepherd NE, Xu W, Lucke AJ, Stoermer MJ, Fairlie DP. Orally absorbed cyclic peptides. Chem Rev 2017; 117: 8094-8128.
| Crossref | Google Scholar | PubMed |
4 Brayden DJ, Hill TA, Fairlie DP, Maher S, Mrsny RJ. Systemic delivery of peptides by the oral route: formulation and medicinal chemistry approaches. Adv Drug Deliv Rev 2020; 157: 2-36.
| Crossref | Google Scholar | PubMed |
5 White TR, Renzelman CM, Rand AC, Rezai T, McEwen CM, Gelev VM, Turner RA, Linington RG, Leung SS, Kalgutkar AS, Bauman JN, Zhang Y, Liras S, Price DA, Mathiowetz AM, Jacobson MP, Lokey RS. On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds. Nat Chem Biol 2011; 7: 810-817.
| Crossref | Google Scholar | PubMed |
6 Bhardwaj G, O’Connor J, Rettie S, Huang YH, Ramelot TA, Mulligan VK, Alpkilic GG, Palmer J, Bera AK, Bick MJ, Di Piazza M, Li X, Hosseinzadeh P, Craven TW, Tejero R, Lauko A, Choi R, Glynn C, Dong L, Griffin R, van Voorhis WC, Rodriguez J, Stewart L, Montelione GT, Craik D, Baker D. Accurate de novo design of membrane-traversing macrocycles. Cell 2022; 185: 3520-3532.e26.
| Crossref | Google Scholar | PubMed |
7 Räder AFB, Reichart F, Weinmüller M, Kessler H. Improving oral bioavailability of cyclic peptides by N-methylation. Bioorg Med Chem 2018; 26: 2766-2773.
| Crossref | Google Scholar |
8 Smith DA, Beaumont K, Maurer TS, Di L. Clearance in drug design. J Med Chem 2019; 62: 2245-2255.
| Crossref | Google Scholar |
9 Smith DA, Beaumont K, Maurer TS, Di L. Relevance of half-life in drug design. J Med Chem 2018; 61: 4273-4282.
| Crossref | Google Scholar | PubMed |
10 Stepan AF, Mascitti V, Beaumont K, Kalgutkar AS. Metabolism-guided drug design. MedChemComm 2013; 4: 631-652.
| Crossref | Google Scholar |
11 Naritomi Y, Terashita S, Kimura S, Suzuki A, Kagayama A, Sugiyama Y. Prediction of human hepatic clearance from in vivo animal experiments and in vitro metabolic studies with liver microsomes from animals and humans. Drug Metab Dispos 2001; 29: 1316-1324.
| Google Scholar | PubMed |
12 Nielsen DS, Lohman R-J, Hoang HN, Fairlie DP, Hill TA. High cell permeability does not predict oral bioavailability for analogues of cyclic heptapeptide sanguinamide A. Aust J Chem 2020; 73: 344-351.
| Crossref | Google Scholar |
13 Boehm M, Beaumont K, Jones R, Kalgutkar AS, Zhang L, Atkinson K, Bai G, Brown JA, Eng H, Goetz GH, Holder BR, Khunte B, Lazzaro S, Limberakis C, Ryu S, Shapiro MJ, Tylaska L, Yan J, Turner R, Leung S, Ramaseshan M, Price DA, Liras S, Jacobson MP, Earp DJ, Lokey RS, Mathiowetz AM, Menhaji-Klotz E. Discovery of potent and orally bioavailable macrocyclic peptide-peptoid hybrid CXCR7 modulators. J Med Chem 2017; 60: 9653-9663.
| Crossref | Google Scholar | PubMed |
14 Lewis I, Schaefer M, Wagner T, Oberer L, Sager E, Wipfli P, Vorherr T. A detailed investigation on conformation, permeability and PK properties of two related cyclohexapeptides. Int J Pept Res Ther 2015; 21: 205-221.
| Crossref | Google Scholar |
15 Rezai T, Yu B, Millhauser GL, Jacobson MP, Lokey RS. Testing the conformational hypothesis of passive membrane permeability using synthetic cyclic peptide diastereomers. J Am Chem Soc 2006; 128: 2510-2511.
| Crossref | Google Scholar | PubMed |
16 Rand AC, Leung SS, Eng H, Rotter CJ, Sharma R, Kalgutkar AS, Zhang Y, Varma MV, Farley KA, Khunte B, Limberakis C, Price DA, Liras S, Mathiowetz AM, Jacobson MP, Lokey RS. Optimizing PK properties of cyclic peptides: the effect of side chain substitutions on permeability and clearance. MedChemComm 2012; 3: 1282-1289.
| Crossref | Google Scholar | PubMed |
17 Wang CK, Northfield SE, Colless B, Chaousis S, Hamernig I, Lohman RJ, Nielsen DS, Schroeder CI, Liras S, Price DA, Fairlie DP, Craik DJ. Rational design and synthesis of an orally bioavailable peptide guided by NMR amide temperature coefficients. Proc Natl Acad Sci USA 2014; 111: 17504-17509.
| Crossref | Google Scholar | PubMed |
18 Wang CK, Northfield SE, Swedberg JE, Colless B, Chaousis S, Price DA, Liras S, Craik DJ. Exploring experimental and computational markers of cyclic peptides: charting islands of permeability. Eur J Med Chem 2015; 97: 202-213.
| Crossref | Google Scholar | PubMed |
19 Nielsen DS, Lohman RJ, Hoang HN, Hill TA, Jones A, Lucke AJ, Fairlie DP. Flexibility versus rigidity for orally bioavailable cyclic hexapeptides. ChemBioChem 2015; 16: 2289-2293.
| Crossref | Google Scholar | PubMed |
20 Hoang HN, Hill TA, Fairlie DP. Connecting hydrophobic surfaces in cyclic peptides increases membrane permeability. Angew Chem Int Ed Engl 2021; 60: 8385-8390.
| Crossref | Google Scholar | PubMed |
21 Vorherr T, Lewis I, Berghausen J, Desrayaud S, Schaefer M. Modulation of oral bioavailability and metabolism for closely related cyclic hexapeptides. Int J Pept Res Ther 2018; 24: 35-48.
| Crossref | Google Scholar | PubMed |
22 Lohman R-J, Nielsen DS, Kok WM, Hoang HN, Hill TA, Fairlie DP. Mirror image pairs of cyclic hexapeptides have different oral bioavailabilities and metabolic stabilities. Chem Commun 2019; 55: 13362-13365.
| Crossref | Google Scholar | PubMed |
23 Hosono Y, Morimoto J, Sando S. A comprehensive study on the effect of backbone stereochemistry of a cyclic hexapeptide on membrane permeability and microsomal stability. Org Biomol Chem 2021; 19: 10326-10331.
| Crossref | Google Scholar | PubMed |
24 Mokkawes T, de Visser SP. Caffeine biodegradation by cytochrome P450 1A2. What determines the product distributions? Chemistry 2023; 29: e202203875.
| Crossref | Google Scholar | PubMed |
25 Gray ALH, Steren CA, Haynes IW, Bermejo GA, Favretto F, Zweckstetter M, Do TD. Structural flexibility of cyclosporine A Is mediated by amide cis–trans isomerization and the chameleonic roles of calcium. J Phys Chem B 2021; 125(4): 1378-1391.
| Crossref | Google Scholar |
26 Ohta K, Agematu H, Yamada T, Kaneko K, Tsuchida T. Production of human metabolites of cyclosporin A, AM1, AM4N and AM9, by microbial conversion. J Biosci Bioeng 2005; 99(4): 390-395.
| Crossref | Google Scholar | PubMed |
27 Bauer J, Spanton S, Henry R, Quick J, Dziki W, Porter W, Morris J. Ritonavir: an extraordinary example of conformational polymorphism. Pharm Res 2001; 18: 859-866.
| Crossref | Google Scholar | PubMed |
28 Sevrioukova IF, Poulos TL. Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc Natl Acad Sci USA 2010; 107: 18422-18427.
| Crossref | Google Scholar | PubMed |
29 Perrin L, Loiseau N, André F, Delaforge M. Metabolism of N-methyl-amide by cytochrome P450s. FEBS J 2011; 278: 2167-2178.
| Crossref | Google Scholar | PubMed |
30 Delaforge M, Andre F, Jaouen M, Dolgos H, Benech H, Gomis JM, Noel JP, Cavelier F, Verducci J, Aubagnac JL, Liebermann B. Metabolism of tentoxin by hepatic cytochrome P-450 3A isozymes. Eur J Biochem 1997; 250: 150-157.
| Crossref | Google Scholar | PubMed |
31 OECD/OCDE Test Guideline No. 117 Partition Coefficient (n-octanol/water), HPLC Method. OECD; 2022. Available at https://www.oecd.org/content/dam/oecd/en/publications/reports/2022/06/test-no-117-partition-coefficient-n-octanol-water-hplc-method_g1gh28e7/9789264069824-en.pdf